 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular crystals of 2-amino-1,3,4-thiadiazole with inorganic oxyacids – crystal engineering, phase transformations and NLO properties†
I.
Matulková
*ab,
J.
Cihelka
a,
M.
Pojarová
b,
K.
Fejfarová
b,
M.
Dušek
b,
I.
Císařová
a,
P.
Vaněk
b,
J.
Kroupa
b,
P.
Němec
c,
N.
Tesařová
c and
I.
Němec
a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, 128 40 Prague 2, Czech Republic. E-mail: matulkov@natur.cuni.cz; Fax: +420 221 951 253; Tel: +420 221 951 247
bInstitute of Physics ASCR, v.v.i., Na Slovance 2, 182 21 Prague 8, Czech Republic
cDepartment of Chemical Physics and Optics, Faculty of Mathematics and Physics, Charles University in Prague, Ke Karlovu 3, 121 16 Prague 2, Czech Republic
First published on 12th December 2013
Abstract
Eight inorganic salts of 2-amino-1,3,4-thiadiazole (2-athd) with hydrochloric, perchloric, nitric, sulphuric, selenic, phosphorous and phosphoric acids were prepared, and their crystal structures were determined. Seven of the compounds crystallise in monoclinic space groups – i.e., 2-athd(1+) chloride monohydrate (P21/c), 2-athd(1+) selenate monohydrate (Pc), 2-athd(1+) dihydrogen phosphate (Cc), 2-athd(1+) phosphite (P21/a), 2-athd(1+) nitrate (P21/c), 2-athd(1+) 2-athd perchlorate (P21/n) and 2-athd(1+) perchlorate (P21/c) – and one crystallises in an orthorhombic space group – 2-athd(1+) hydrogen sulphate (P212121). The thermal behaviours of the materials were studied (DSC) down to liquid nitrogen temperature, and the properties of the perchlorate, hydrogen sulphate and selenate monohydrate crystals were discussed with respect to the results of high or low-temperature X-ray diffraction, IR and Raman spectroscopic studies. Quantitative measurements of the second harmonic generation of the powdered non-centrosymmetric samples at 1064 and 800 nm were performed and a relative efficiency compared to KDP is presented.
Introduction
Crystal engineering represents one of the most promising fields of supramolecular chemistry and addresses the design of novel materials with desired properties.1–3 The formation of crystals from molecules and ions is a very difficult process that depends not only on the symmetry of the molecules involved but also on the intermolecular (supramolecular) interactions, which direct the mutual assembly of the building blocks. Out of these interactions with different natures4 (i.e., ion–ion interactions, ion–dipole interactions, dipole–dipole interactions, cation–π interactions, anion–π interactions, π–π interactions and van der Waals forces), hydrogen bonding, which is sometimes classified as a dipole–dipole interaction, can be considered as the most important interaction (especially in the preparation of materials for non-linear optics).Nonlinear optics (NLO) is the branch of science that describes the interaction of light with matter where the principle of linear superposition is ignored. Examples of non-linear optical interactions include harmonic generation (especially second harmonic generation), sum- and difference-frequency generation, intensity dependence of the complex refractive index, light-by-light scattering, and stimulated light scattering.1 These properties lead to many applications, such as all-optical switching, optical power limiting, image manipulation, image processing, and data storage.5
The materials used for second harmonic generation (SHG) can be divided into three principle groups based on their chemical natures.6 The first group (also of historical consequences) is formed by inorganic salts and oxides. These inorganic compounds are currently the most utilised materials in the above-mentioned technical applications. The second group, which is formed by organic compounds, is a very promising class due to their larger hyperpolarisability values, higher resistance to optical damage and the possibilities for molecular design.2 A limiting factor for the applications of these organic materials (usually exhibiting a high dipole moment) in the crystalline state is their tendency to form symmetric pairs, which leads to a centrosymmetric arrangement of the crystal structures and, consequently, the exclusion of SHG. The third group of materials is closely related to the second one and is based on hydrogen-bonded salts or cocrystals of suitable polarisable organic molecules, which are mainly responsible for NLO properties. The energy of the hydrogen bonds can counteract the tendencies of organic molecules and ions with highly delocalised π-electron system to form centrosymmetric pairs. In addition, the hydrogen-bonded structures frequently exhibit advantageous chemical and physical properties.3,7
Recently, some authors reported8 the synthesis and characterisation of new chromophores, where the replacement of the benzene ring by less aromatic heterocycles (especially derivatives of thiazole) leads to significant bathochromic shift and to larger molecular hyperpolarisability. The present paper, which follows our previous investigation of triazolium compounds,9–12 is focused on crystal engineering of novel materials for nonlinear optics based on organic sulphur-containing heteroaromatic molecules. The selection of the 2-amino-1,3,4-thiadiazole molecule was motivated by the fact that the calculated first hyperpolarisability values (see Second harmonic generation section) are a factor of ten larger than those for the studied aminotriazoles11 and aminothiazoles.13
2-Amino-1,3,4-thiadiazole and its derivatives have also been investigated in medicine as very promising molecules for the treatment of cancer14 and as receptor antagonists.15
In this paper, we have prepared eight novel inorganic salts of 2-amino-1,3,4-thiadiazole: 2-amino-1,3,4-thiadiazolium(1+) chloride monohydrate (2-athdClH2O), 2-amino-1,3,4-thiadiazolium(1+) selenate monohydrate (2-athd2SeO4H2O), 2-amino-1,3,4-thiadiazolium(1+) dihydrogen phosphate (2-athdH2PO4), 2-amino-1,3,4-thiadiazolium(1+) phosphate (2-athd2HPO3), 2-amino-1,3,4-thiadiazolium(1+) nitrate (2-athdNO3), 2-amino-1,3,4-thiadiazolium(1+) 2-amino-1,3,4-thiadiazole perchlorate (2-athd2ClO4), 2-amino-1,3,4-thiadiazolium(1+) perchlorate (2-athdClO4) and 2-amino-1,3,4-thiadiazolium(1+) hydrogen sulphate (2-athdHSO4). All salts were studied using single crystal and powder X-ray diffraction, vibrational spectroscopy and differential scanning calorimetry. The thermal effects observed in 2-athd2ClO4, 2-athdClO4, 2-athdHSO4 and 2-athd2SeO4H2O crystals were also discussed. The non-centrosymmetric salts 2-athdHSO4, 2-athd2SeO4H2O and 2-athdH2PO4 were found to be promising materials for second harmonic generation (SHG).
Experimental
Crystals of all eight new compounds of 2-amino-1,3,4-thiadiazolium(1+) were prepared by the addition of an aqueous solution of the pertinent inorganic acid to a solution of 2-amino-1,3,4-thiadiazole in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The solutions (colourless or slightly yellow) were stored in the dark at room temperature. The resulting crystals were filtered off and dried in air. In the 2-amino-1,3,4-thiadiazole and perchloric acid solutions, two types of crystals were obtained from the 1:1 molar ratio – 2-athd2ClO4 (needles) and 2-athdClO4 (plates); at a 2:1 molar ratio, only 2-athd2ClO4 crystallised.
:1 molar ratio. The solutions (colourless or slightly yellow) were stored in the dark at room temperature. The resulting crystals were filtered off and dried in air. In the 2-amino-1,3,4-thiadiazole and perchloric acid solutions, two types of crystals were obtained from the 1:1 molar ratio – 2-athd2ClO4 (needles) and 2-athdClO4 (plates); at a 2:1 molar ratio, only 2-athd2ClO4 crystallised.
Single-crystal X-ray diffraction data of 2-athdClH2O, 2-athdNO3, 2-athd2ClO4, 2-athdHSO4, 2-athd2SeO4H2O, 2-athd2HPO3 and 2-athdH2PO4 were collected at 120 K on an Oxford Diffraction four-cycle diffractometer Gemini using mirror-collimated CuKα radiation and an Atlas CCD detector. Single-crystal X-ray diffraction temperature-dependent data of 2-athdClO4 and 2-athdHSO4 (temperature interval from 290 K to 110 K with a step size of 20 K) and of 2-athd2ClO4 (three temperatures of 150, 283 and 315 K) were collected on a Nonius Kappa CCD diffractometer (MoKα radiation, graphite monochromator and a Bruker APEX-II CCD detector). The temperature was controlled using an Oxford Cryosystems liquid nitrogen cryostream cooler. The phase problem was solved with direct methods by the computer program SIR2002.16 The non-hydrogen atoms were refined anisotropically using the full-matrix least-squares procedure by the programs SHELXL97,17 SHELXS,18 SIR9219 or CRYSTALS.20 In most cases, the hydrogen atoms were found in the Fourier difference maps, and the hydrogen atoms attached to the carbon atoms were repositioned geometrically. The positions of the hydrogen atoms attached to nitrogen were refined in the initial stages but were usually fixed to travel with the nitrogen atom. The positions of the hydrogen atoms of water molecules were refined with a distance restraint. The isotropic temperature parameters of the hydrogen atoms were calculated as 1.2–1.5 Ueq of the parent atom. In 2-athd2SeO4H2O, the structural analysis was complicated by the poor quality of the single crystals. Thus, the hydrogen atoms of the water molecule could not be determined for this compound, and the thermal parameters of the oxygen atoms in the selenate anion had to be restrained to be identical. Further details of the particular refinements can be found in the CIF files and ESI† materials.
The basic crystallographic data, measurement and refinement details are summarised in Tables 1 and 2. The crystallographic data for 2-athdClH2O, 2-athdNO3, 2-athd2ClO4, 2-athdClO4, 2-athdHSO4, 2-athd2SeO4H2O, 2-athd2HPO3 and 2-athdH2PO4 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 910449, CCDC 910447, CCDC 910450, CCDC 910853, CCDC 910445, CCDC 910448, CCDC 910444 and CCDC 910446, respectively.
| Identification code | 2-athdNO3 | 2-athdClH2O | 2-athd2ClO4 | 2-athdClO4 | ||||
|---|---|---|---|---|---|---|---|---|
| Empirical formula | C2H4N4O3S | C2H6ClN3OS | C4H7ClN6O4S2 | C2H4ClN3O4S | ||||
| Formula weight | 164.15 | 155.61 | 302.73 | 201.6 | ||||
| Temperature | 120(2) K | 120(2) K | 120(2) K | 150(2) K | ||||
| a | 6.6897(2) Å | 4.9574(1) Å | 4.9817(1) Å | 5.0168(3) Å | ||||
| b | 8.3773(2) Å | 8.7771(2) Å | 20.0865(3) Å | 18.6658(11) Å | ||||
| c | 10.9734(3) Å | 14.2055(3) Å | 11.0569(2) Å | 7.4367(5) Å | ||||
| α | 90° | 90° | 90° | 90° | ||||
| β | 103.301(3)° | 97.481(2)° | 90.643(2)° | 102.148(2)° | ||||
| γ | 90° | 90° | 90° | 90° | ||||
| Volume | 598.47(3) Å3 | 612.84(2) Å3 | 1106.34(3) Å3 | 680.80(7) Å3 | ||||
| Z | 4 | 4 | 4 | 4 | ||||
| Calculated density | 1.822 Mg m−3 | 1.687 Mg m−3 | 1.817 Mg m−3 | 1.967 Mg m−3 | ||||
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | ||||
| Space group | P21/c | P21/c | P21/n | P21/c | ||||
| Absorption coefficient | 4.516 mm−1 | 7.974 mm−1 | 6.797 mm−1 | 0.842 mm−1 | ||||
| F(000) | 336 | 320 | 616 | 420 | ||||
| Crystal size | 0.47 × 0.31 × 0.10 mm | 0.47 × 0.09 × 0.06 mm | 0.61 × 0.16 × 0.13 mm | 0.56 × 0.33 × 0.19 mm | ||||
| Diffractometer and radiation | Four-cycle diffractometer, Cu λ = 1.54184 Å | Nonius Kappa CCD, Mo λ = 0.71073 Å | ||||||
| Completeness to θ | 62.43 | 98.3% | 62.58 | 97.8% | 62.61 | 98.5% | 30.00 | 99.7% |
| Range of h, k and l | −7→ 7, −9→9, −12→12 |
−5→ 5, −9→10, −16→15 |
−5→ 4, −21→22, −12→12 |
−7→ 6, −20→26, −8→10 |
||||
| θ range for data collection | 6.72 to 62.32° | 5.94 to 62.58° | 4.00 to 62.52° | 2.18 to 30.00° | ||||
| Reflection collected/unique (Rint) | 4238/935 (0.0153) | 2330/961 (0.0369) | 4310/1734 (0.0128) | 5630/1979 (0.0135) | ||||
| No. of observed reflection | 841 | 913 | 1686 | 1870 | ||||
| Criterion for observed reflection | I > 2σ(I) | I > 2σ(I) | I > 2σ(I) | I > 2σ(I) | ||||
| Absorption correction | Multi-scan | Analytical | Multi-scan | None | ||||
| Function minimized | ∑w(Fo2 − Fc2)2 | ∑w(Fo2 − Fc2)2 | ∑w(Fo2 − Fc2)2 | ∑w(Fo2 − Fc2)2 | ||||
| Parameters refined | 91 | 73 | 155 | 104 | ||||
| R, wR [I > 2σ(I)] | 0.0435; 0.0402 | 0.0652; 0.1810 | 0.0301; 0.0793 | 0.0223; 0.0646 | ||||
| R, wR (all data) | 0.1125; 0.1077 | 0.0642; 0.1793 | 0.0292; 0.0784 | 0.0235; 0.0662 | ||||
| Value of S | 1.099 | 1.131 | 1.037 | 0.597 | ||||
| Max and min. heights in final Δρ map | 0.442 and −0.383 e Å−3 | 1.343 and −0.613 e Å−3 | 0.720 and −0.373 e Å−3 | 0.322 and −0.471 e Å−3 | ||||
| Weighting scheme | w = 1/[σ2Fo2 + (aP)2 + bP] | P = (Fo2 + 2Fc2)/3 | ||||||
| a = 0.0752 | a = 0.01422 | a = 0.0466 | a = 0.0672 | |||||
| b = 0.4177 | b = 0.2885 | b = 1.2065 | b = 1.1778 | |||||
| Sources of atomic scattering factors | SHELXL9717 | |||||||
| Programs used | SHELXL97,17 PLATON,24 SIR9219 | |||||||
| Identification code | 2-athd2HPO3 | 2-athdH2PO4 | 2-athd2SeO4H2O | 2-athdHSO4 | ||||
|---|---|---|---|---|---|---|---|---|
| Empirical formula | C2H5N3O3PS | C2H6N3O4PS | C4H8N6O5S2Se | C2H5N3O4S2 | ||||
| Formula weight | 182.12 | 199.13 | 363.26 | 199.21 | ||||
| Temperature | 120(2) K | 120(2) K | 120(2) K | 120(2) K | ||||
| a | 6.2442(5) Å | 4.4119(1) Å | 5.5377(1) Å | 5.20000(7) Å | ||||
| b | 13.1523(12) Å | 21.9024(5) Å | 13.9715(2) Å | 10.22430(15) Å | ||||
| c | 8.5929(5) Å | 7.5190(2) Å | 7.6737(1)Å | 13.06242(17) Å | ||||
| α | 90° | 90° | 90° | 90° | ||||
| β | 96.820(6)° | 94.531(2)° | 93.472(1)° | 90° | ||||
| γ | 90° | 90° | 90° | 90° | ||||
| Volume | 700.70(9) Å3 | 724.30(3) Å3 | 592.62(2) Å3 | 694.480(18) Å3 | ||||
| Z | 4 | 4 | 2 | 4 | ||||
| Calculated density | 1.726 Mg m−3 | 1.826 Mg m−3 | 2.036 Mg m−3 | 1.905 Mg m−3 | ||||
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | ||||
| Space group | P21/a | Cc | Pc | P212121 | ||||
| Absorption coefficient | 5.973 mm−1 | 1.826 mm−1 | 7.885 mm−1 | 6.830 mm−1 | ||||
| F(000) | 372 | 408 | 360 | 408 | ||||
| Crystal size | 0.40 × 0.22 × 0.13 mm | 0.44 × 0.35 × 0.08 mm | 0.59 × 0.44 × 0.13 mm | 0.45 × 0.32 × 0.15 mm | ||||
| Diffractometer and radiation | Four-cycle diffractometer, Cu λ = 1.54184 Å | |||||||
| Completeness to θ | 61.81 | 99.6% | 62.42 | 97.9% | 62.47 | 98.6% | 62.47 | 99.9% |
| Range of h, k and l | −7→ 6, −15→14, −9→ 9 |
−4→4 , −24→24, −8→ 7 |
−6→ 6, −15→15, −8→ 8 |
−5→ 5, −11→11, −14→15 |
||||
| θ range for data collection | 3.36 to 62.98° | 4.03 to 62.33° | 3.16 to 62.47° | 5.49 to 62.47° | ||||
| Reflection collected/unique (Rint) | 5609/1123 (0.061) | 2156/874 (0.0373) | 2741/1274 (0.0293) | 8266/1094 (0.0222) | ||||
| No. of observed reflection | 962 | 872 | 1274 | 1094 | ||||
| Criterion for observed reflection | I > 2σ(I) | I > 2σ(I) | I > 2σ(I) | I > 2σ(I) | ||||
| Absorption correction | Analytical | Multi-scan | Multi-scan | Analytical | ||||
| Function minimized | F | ∑w(Fo2 − Fc2)2 | ∑w(Fo2 − Fc2)2 | ∑w(Fo2 − Fc2)2 | ||||
| Parameters refined | 962 | 102 | 141 | 121 | ||||
| R, wR [I > 3σ(I)] | 0.0769; 0.0662 | 0.0297; 0.0840 | 0.0350; 0.0873 | 0.0202; 0.0202 | ||||
| R, wR (all data) | 0.0834; 0.0679 | 0.0296; 0.0839 | 0.0350; 0.0873 | 0.0503; 0.0503 | ||||
| Value of S | 1.1054 | 1.080 | 1.198 | 0.879 | ||||
| Max and min. heights in final Δρ map | 1.11 and −0.63 e Å−3 | 0.37 and −0.34 e Å−3 | 0.524 and −1.247 e Å−3 | 0.303 and −0.280 e Å−3 | ||||
| Weighting scheme | w = 1/[A0T0(x) + A1T1(x)+…+An−1Tn−1(x)] | w = 1/[σ2Fo2 + (bP)2 + bP] | P = (Fo2 + 2Fc2)/3 | |||||
| x = Fcalc/Fmax | a = 0.0618 | a = 0.0680 | a = 0.0221 | |||||
| A i = 24.3; 24.3; 22.8; 10.0 | b = 0.7109 | b = 0.0511 | b = 1.1901 | |||||
| Sources of atomic scattering factors | CRYSTALS20 | SHELXL9717 | ||||||
| Programs used | CRYSTALS,20 PLATON24 | SHELXL97,17 PLATON,24 SIR9219 | ||||||
The phase purity of the prepared polycrystalline samples was also controlled in the powder X-ray diffraction at room temperature using the Bragg–Brentano geometry on a Philips X'pert PRO MPD X-ray diffraction system equipped with an ultrafast X'Celerator detector using a Cu-anode (Cu-Kα; λ = 1.5418 Å). The recorded diffraction patterns (see Table 1S–8S; ESI†) are consistent with the calculated diffraction maxima (FullProf software21).
The temperature-dependent FTIR and FT Raman measurements were carried out on a Nicolet 6700 FTIR spectrometer (resolution 2 cm−1, Happ–Genzel apodisation) equipped with a Nexus FT Raman module using the Oxford Instruments' liquid nitrogen cryostat Optistat DN-V (KRS-5 windows for mid IR, i.e., 4000–400 cm−1 region, polypropylene windows for far IR, i.e., 600–50 cm−1 region, and quartz window for FT Raman, i.e., 3700–150 cm−1 region) in the 298–77 K interval. Low-temperature vibrational spectra of 2-athd2SeO4H2O, 2-athdHSO4 and 2-athdClO4 were measured from 77 K to 298 K with a step size of 20 K. A temperature step of 10 K was selected in the regions close to the thermal effects observed in DSC curves. The vibrational spectra of 2-athd2ClO4 were recorded from a temperature of 290 K to 340 K with a step size of 10 K. A temperature step of 5 K was selected near the recorded DSC effect. The temperature was controlled using an ITC 503S controller with a precision of ±0.1 K. The Nujol mull and polyethylene pellet methods were used for the recording of the spectra in the mid and far IR regions, respectively.
The DSC measurements of all the compounds were carried out on Perkin Elmer Pyris Diamond DSC and DSC 7 instruments in the 93–473 K temperature region (nitrogen and helium atmospheres above and below 298 K, respectively – 20 ml min−1). A heating rate of 10 K min−1 was selected to measure approximately 20 mg of the finely ground sample placed in hermetically sealed aluminium pans.
The UV-Vis-NIR spectra of aqueous solutions of all the prepared salts were recorded in the 190–800 nm range using a Perkin Elmer Lambda 19 UV-Vis-NIR spectrometer.
The quantum chemical calculations (GAUSSIAN09W22) of 2-amino-1,3,4-thiadiazole and 2-amino-1,3,4-thiadiazolium(1+) cation were performed using the closed-shell restricted density functional method (B3LYP) with the 6-311+G(d,p) basis set, applying tight convergence criteria and an ultrafine integration grid. The visualisation of the results was carried out with the GaussView23 program package. The geometry optimisations, also yielding the molecular energies, were followed by the first hyperpolarisability component calculations. The calculated geometries of 2-amino-1,3,4-thiadiazole (point group C1; E = −640.54 Hartree) and 2-amino-1,3,4-thiadiazolium(1+) cation (point group C1; E = −640.98 Hartree) were compared with those determined by X-ray structure analysis.
SHG measurements were performed using a Q-switched Nd-YAG laser (6 ns pulses at 20 Hz, λ = 1064 nm). For the quantitative determination of the SHG efficiency, the intensity of the transmitted laser light at 532 nm generated in the sample was measured using a photomultiplier and a boxcar average, and the signal was compared with the one generated in KDP. Very weak pulses (3 mJ) were used to avoid sample damage. The experiment was performed on powdered samples (75–150 μm particle size) placed between two glass plates, and the measurements were repeated on different areas of the same sample (the results were averaged).
The additional measurements of SHG at 800 nm were carried out with 160 fs laser pulses generated at an 82 MHz repetition rate by a Ti:sapphire laser (MaiTai, Spectra Physics). For the quantitative determination of the SHG efficiency, the intensity of the back-scattered laser light at 400 nm generated in the sample was measured using a grating spectrograph with a diode array (InstaSpec II, Oriel), and the signal was compared with the one generated in KDP (i.e., KH2PO4). The experiment was performed using powdered samples (75–150 μm particle size) loaded into 5 mm glass cells with the aid of a vibrator, and the measurements were repeated on different areas of the same sample (the results were averaged). This experimental procedure minimises the signal fluctuations induced by sample packing.
Results and discussion
Crystal structures
The atom numbering of 2-amino-1,3,4-thiadiazole and 2-amino-1,3,4-thiadiazolium(1+) cation in the title structures is given in Fig. 1S–8S (ESI;† PLATON24 software). The selected bond lengths and angles, including those of the hydrogen bonds, are listed in Tables 9S–16S (ESI†).In the crystal structures of the title compounds and pure 2-amino-1,3,4-thiadiazole,25 the heteroaromatic rings are planar. The aromaticity of this ring is reflected in the values of C–N, N–N and N–S bond lengths as shown in Tables 9S–16S (ESI†).
The 2-athd(1+) cations are interconnected in centrosymmetric pairs (2-athd2ClO4 and 2-athdClH2O) or in chains (2-athd2SeO4H2O). The interactions between the cations can be described by the graph set descriptors26 R22(7) or R22(6) for pairs in 2-athd2ClO4 or 2-athdClH2O, respectively. The graph set descriptors C(5) represent the chains in the structure of 2-athd2SeO4H2O. In all structures, the hydrogen bonds of N–H⋯N type connecting the 2-athd(1+) cations (or the cation and molecule of 2-athd in the case of 2-athd2ClO4) are formed by H-donor atoms from the NH2 or NH groups, and the ring nitrogens act as H-acceptors (N4, N24 and N23 in the case of 2-athd2ClO4). A detailed comparison of the results is presented in the corresponding structural descriptions in the following paragraphs.
The layered structure of 2-athdNO3 is formed by a common network of nitrate anions and 2-amino-1,3,4-thiadiazolium(1+) cations. The layers are perpendicular to the ac plane, see Fig. 1. The hydrogen bond interactions in the crystal structures can be described by graph set motifs C(3) and C(5).
 | ||
| Fig. 1 Packing scheme of 2-athdNO3 (view along the [010] direction). The dashed lines indicate hydrogen bonds. | ||
The structure of 2-athdClH2O contains centrosymmetric cationic pairs. These cationic pairs fill cavities, which are formed by chains of common interaction between the chloride anion and the water molecule (see Fig. 2). The pairs are situated about the crystallographic inversion centres, and the chains are parallel to the a axis (see Fig. 9S; ESI†). The cationic pairs are connected by hydrogen bonds N3–H3⋯N4 (N3⋯N4 distance: 3.103(4) Å). The pairs are connected via chains formed by chlorine and water molecules in a 3D crystal structure (graph set motifs R12(6), R22(6), C(3) and C(5) – see Fig. 10S; ESI†). The presence of a number of weak N–H⋯Cl interactions (see Fig. 2) is another characteristic feature of this crystal structure.
 | ||
| Fig. 2 Packing scheme of 2-athdClH2O (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The crystal structure of 2-athd2ClO4 contains layers of organic base pairs (formed by an interconnected 2-amino-1,3,4-thiadiazole molecule and its cation) arranged in a 3D structure via N–H⋯O interactions with anions, which are localised between the organic layers. One such layer is depicted in Fig. 3. The layers are parallel to the [1 1 0] direction and perpendicular to the a axis. In each layer, a pair is connected by the hydrogen bonds N3–H3⋯N24 (the N3⋯N24 distance is 2.759(3) Å) and N6–H6A⋯N23 (the N6⋯N23 distance is 2.880(3) Å). This arrangement can be described using the ring and chain graph set descriptors R22(7) and C(3). The connection between the anion and the cationic pair is provided by N–H⋯O interactions ranging from 2.937(2) to 3.052(3) Å.
 | ||
| Fig. 3 Packing scheme of 2-athd2ClO4 (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The crystal structure of 2-athdClO4 contains a 3D common network of 2-amino-1,3,4-thiadiazolium(1+) cations and perchlorate anions. The structure is connected by the hydrogen bonds of N–H⋯O type with the donor⋯acceptor distances ranging from 2.945(1) Å to 3.089(1) Å and by weak C–H⋯O interactions (C⋯O distance of 3.294(2) Å). This complicated 3D arrangement is very difficult to describe using the ring and chain graph set descriptors, see Fig. 4.
 | ||
| Fig. 4 Packing scheme of 2-athdClO4 (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The structure of 2-athd2HPO3 (see Fig. 5) consists of layers of alternating cations and anions parallel to the ac plane. All phosphite anions located in one layer are oriented with the P–H bond out of the layer plane and the neighbouring layers form centrosymmetric pairs. The entire 3D crystal structure is stabilised by N–H⋯O hydrogen bonds with donor⋯acceptor distances ranging from 2.654(4) to 2.820(4) Å. The arrangement in one layer can be described by the graph set motifs R22(8) and C(6), see Fig. 11S (ESI†).
 | ||
| Fig. 5 Packing scheme of 2-athdHPO3 (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The structure of 2-athdH2PO4 belongs to the monoclinic system with a non-centrosymmetric Cc space group, which is very promising for subsequent studies of the NLO properties, especially SHG. The structure is formed with the anionic layers (see Fig. 12S; ESI†) parallel to the ac plane, graph set motifs R44(16) and C(5), which are interconnected by the cations in a 3D network, graph set motifs R23(10) – see Fig. 6.
 | ||
| Fig. 6 Packing scheme of 2-athdH2PO4 (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The non-centrosymmetric crystal of 2-athd2SeO4H2O belongs to the monoclinic space group Pc. The asymmetric unit consists of two 2-athd(1+) cations, a selenate anion and a water molecule. One of the cations forms the chains through N26–H26B⋯N24 hydrogen bonds with a donor⋯acceptor distance of 3.006(6) Å. The second type of 2-athd(1+) cation does not interact with the other cation, but the conformation is stabilised by π–π stacking with a centroid distance of 3.837(2) Å to build the columns. The chains of 2-athd(1+) cations and columns of stacked 2-athd(1+) cations are interconnected by N–H⋯O interactions with the selenate anions. The crystal structure can be described by the graph set descriptors R32(7), R22(8) and C(5), see Fig. 7.
 | ||
| Fig. 7 Packing scheme of 2-athd2SeO4H2O (view along the [100] direction). The dashed lines indicate hydrogen bonds. | ||
The last non-centrosymmetric crystal structure of 2-athdHSO4 is orthorhombic with the space group P212121. The crystal structure is based on the anionic chains formed by O4–H7⋯O3 hydrogen bonds with a donor⋯acceptor distance equal to 2.620(2) Å. These hydrogen sulphate chains are parallel to the a axis. Each anionic chain is interconnected via four 2-athd(1+) cations with four neighbouring anionic chains. The arrangement of the crystal structure (see Fig. 8) can be described by the graph set descriptors R23(10), C(5) and C(3).
 | ||
| Fig. 8 Packing scheme of 2-athdHSO4 (view along the [010] direction). The dashed lines indicate hydrogen bonds. | ||
From a general point of view, the simplest crystal structure of the title salts is 2-athdNO3, which has just the layer structure with layers perpendicular to the [010] direction. The presence of tetrahedral perchlorate anions in 2-athdClO4 crystals leads to the formation of the 3D structure. The more complicated crystal structure of 2-athd2ClO4 contains pairs formed by the 2-athd molecule and the 2-athd(1+) cation, which are involved in a 3D hydrogen bonding network through the perchlorate anions. The cationic pairs interconnect chains formed by chlorine and water molecules in the 3D crystal structure of 2-athdClH2O. The crystal structures of 2-athdHSO4 and 2-athdH2PO4 are based on anionic chains (formed by hydrogen sulphates) or 3D networks (formed by dihydrogen phosphates), which encapsulate the 2-athd(1+) cations. The remaining two structures, 2-athd2SeO4H2O and 2-athd2HPO3, are formed by networks of alternating cations and anions.
The important geometric parameters for the discussion of the mentioned crystal structures of the inorganic salts of 2-amino-1,3,4-thiadiazolium(1+) and 2-amino-1,3,4-thiadiazole itself are as follows: (1) the angle between the ring plane and the plane defined by the NH2 group, and (2) the distance between the thiadiazole ring plane and the H3 hydrogen atom of the ring N–H bond. The values of both parameters are listed in Table 17S (ESI†) for all title structures.
The amino group is inclined towards the plane of the ring at an angle ranging from 0.3 to 17.3°. The intermediate values, i.e., 8.61(1), 5.29(1) and 4.84(1)°, give the structures of 2-athd2HPO3, 2-athdHSO4 and 2-athd, respectively. The highest values of 17.24(0) and 15.15(1)° were found in the structures of 2-athdH2PO4 and 2-athd2ClO4, respectively. The hydrogen atom attached to the nitrogen atom of the thiadiazolium cation ring (H3) also slightly deviates from the ring plane (the distance of the H3 atom from the ring plane ranges from −0.1465 to 0.003 Å). The positions of the H3 atoms in the structures are significantly influenced by hydrogen bond interactions of the N–H⋯N and N–H⋯O types.
Geometry optimisation of 2-amino-1,3,4-thiadiazole and 2-amino-1,3,4-thiadiazolium(1+)
To calculate the first hyperpolarisability components of 2-amino-1,3,4-thiadiazole and the 2-amino-1,3,4-thiadiazolium(1+) cation, a computational geometry optimisation of the isolated single molecule and its cation were performed. The selection of the density functional methods (B3LYP) and 6-311+G(d,p) basis set in principle results from the comparison of the results obtained from the other method (HF/6-311+G) and the previous computation of this and similar molecules.10,11The geometric parameters obtained for the optimised molecule and the ion are compared with the results of the X-ray structure in Tables 18S–19S (ESI†). The optimised values of the bond lengths and inter-atomic angles are scattered around the average values obtained from the crystal structures. The maximum deviation, 1.2°, was observed for the average value of the C2–N3–N4 (2-athd) and C22–S21–C25 (2-athd(1+)) angles, and the maximum deviation for the bond lengths was 0.105 Å for the C5–S1 bond in 2-athd(1+) and 0.026 Å for the C22–S21 bond in 2-athd.
Thermal behaviour
To evaluate the thermal stability of these materials, DSC measurements were carried out for all of the compounds over a temperature range from 93 K to temperatures near their melting points. The crystals of 2-athdClH2O (m.p. 380 K), 2-athd2ClO4 (m.p. 389 K), 2-athdClO4 (m.p. 399 K), 2-athdNO3 (m.p. 415 K), 2-athdHSO4 (m.p. 385 K), 2-athd2SeO4H2O (m.p. 378 K), 2-athdH2PO4 (m.p. 415 K) and 2-athdHPO3 (m.p. 394 K) are stable in air up to their melting points, which are listed in parentheses. Only the crystals of 2-athd2ClO4, 2-athdClO4, 2-athdHSO4 and 2-athd2SeO4H2O exhibit weak anomalies (effects) on both the heating and cooling runs.The DSC curves of 2-athd2SeO4H2O exhibit only one reproducible anomaly, with the shape characteristic of a “glass transition” (Tg = 191 K, Δcp = 3.2 × 10−2 J g−1 K−1 on the heating run; Tg = 187 K, Δcp = 2.3 × 10−2 J g−1 K−1 on the cooling run; see Fig. 13S; ESI†).
A similar effect was also recorded for 2-athdHSO4 (Tg = 160 K, Δcp = 6.2 × 10−2 J g−1 K−1 on the heating run; Tg = 154 K, Δcp = 7.0 × 10−2 J g−1 K−1 on the cooling run; see Fig. 14S; ESI†). Furthermore, the DSC curves of 2-athdHSO4 contain two reproducible effects observed only on the heating runs: an exothermic peak (Tt = 212 K, ΔH = −1.57 J g−1) and an endothermic, λ-shaped peak (Tt = 236 K, ΔH = 0.99 J g−1).
The crystals of 2-athdClO4 exhibit a λ-shaped anomaly with considerable thermal hysteresis (see Fig. 15S; ESI†), which is also associated with the splitting of the peak on the cooling run (Tt = 271 K, ΔH = 0.62 J g−1 on the heating run; two peaks on the cooling run at Tt = 241 and 231 K, ΔH = −0.37 and −1.04 J g−1, respectively).
The DSC curves recorded for the 2-athd2ClO4 crystals contain a reproducible, wide anomaly slightly above room temperature (Tt = 303 K, ΔH = 2.86 J g−1 on the heating run; Tt = 301 K, ΔH = −3.36 J g−1 on the cooling run; see Fig. 16S; ESI†).
A temperature-dependent X-ray structural analysis and a vibrational spectroscopic study were consequently performed to explain the nature of the observed thermal effects. The low-temperature diffraction data collection and structure determination of 2-athdHSO4 and 2-athdClO4 crystals at temperatures above and below the observed thermal effects were performed in the first step. Unfortunately, we were not able to repeat this procedure for the 2-athd2SeO4H2O single crystal, which always cracked during the cooling. Because only subtle changes in the low-temperature crystal structures were observed for 2-athdHSO4 and 2-athdClO4, we consequently performed detailed X-ray structural analyses after cooling the samples from 290 K to 110 K with a step size of 20 K. The unit cell parameters and crystal structures for both salts were determined for every temperature step. The temperature-dependent variation in the selected unit cell parameters a, b, c and β, see Fig. 9, indicates very subtle changes in the structures of 2-athdClO4 and 2-athdHSO4.
 | ||
| Fig. 9 Temperature dependence of lattice parameters: (a) parameter b of 2-athdHSO4; (b) parameter c of 2-athdHSO4; (c) parameter b of 2-athdClO4; (d) parameter β of 2-athdClO4. The uncertainty is indicated by the intercept for each point in the graph. The dashed lines represent the different slopes of smooth data. The arrows indicate the outlying points. | ||
The values of the unit cell (space group P212121) parameters (a, b, c and V) decrease with decreasing temperature in the 2-athdHSO4 structure (see Table 3). The observed continuous changes can be depicted by the values of Δa/a = 0.94%, Δb/b = 0.38%, Δc/c = 0.67% and ΔV/V = 1.84%. Because the distortion (i.e., thermal expansion/contraction) of the crystal can be described by the change in the volume expansion/contraction coefficient defined in a previous paper,27 we have calculated (see Table 4) the principal linear thermal expansion/contraction coefficients along the crystallographic axes for the temperature changes in the 2-athdHSO4 crystal. It is apparent that the studied temperature interval can be expressed by one set of calculated coefficients. Only three small discontinuities in the temperature dependencies of the unit cell parameters were observed at the temperatures of 110, 190 and 230 K (see Fig. 9a and b). Observed discontinuities can be very roughly correlated with the presence of thermal effects in DSC curves at Tg = 160 K, Tt = 212 K and Tt = 236 K.
| Unit cell parametrs of 2-athdHSO4 | ||||
|---|---|---|---|---|
| Temperature (K) | a (Å) | b (Å) | c (Å) | V (Å3) |
| 110 | 5.1991(3) | 10.2216(6) | 13.0731(8) | 694.75(7) |
| 130 | 5.2028(2) | 10.2263(5) | 13.0701(6) | 695.40(5) |
| 150 | 5.2094(1) | 10.2327(3) | 13.0826(4) | 697.38(3) |
| 170 | 5.2141(2) | 10.2361(3) | 13.0916(4) | 698.73(4) |
| 190 | 5.2199(2) | 10.2372(4) | 13.1048(5) | 700.28(5) |
| 210 | 5.2252(1) | 10.2447(3) | 13.1173(4) | 702.18(3) |
| 230 | 5.2292(2) | 10.2435(3) | 13.1171(4) | 702.62(4) |
| 250 | 5.2367(1) | 10.2532(3) | 13.1402(4) | 705.54(3) |
| 270 | 5.2441(2) | 10.2577(3) | 13.1500(5) | 707.37(4) |
| 290 | 5.2483(1) | 10.2607(3) | 13.1614(4) | 708.76(3) |
| Unit cell parameter | Coefficient | 2-athdHSO4 | 2-athdClO4 | |
|---|---|---|---|---|
| 110–290 K | 110–200 K | 200–290 K | ||
| a | a 0 (Å) | 5.1669 | 4.9628 | 4.9513 |
| μ a (Å K−1) | 2.80 × 10−4 | 3.44 × 10−4 | 4.00 × 10−4 | |
| α a (K−1) | 5.34 × 10−5 | 6.79 × 10−5 | 7.89 × 10−5 | |
| b | b 0 (Å) | 10.2009 | 18.5785 | |
| μ b (Å K−1) | 2.05 × 10−4 | 6.67 × 10−4 | ||
| α b (K−1) | 2.00 × 10−5 | 3.55 × 10−5 | ||
| c | c 0 (Å) | 13.0008 | 7.4125 | 7.3852 |
| μ c (Å K−1) | 5.46 × 10−4 | 1.48 × 10−4 | 3.02 × 10−4 | |
| α c (K−1) | 2.77 × 10−5 | 1.98 × 10−5 | 4.04 × 10−5 | |
| β | β 0 (°) | 102.527 | 102.753 | |
| μ β (K−1) | −2.68 × 10−3 | −3.78 × 10−3 | ||
| α β (K−1) | −2.64 × 10−5 | −3.72 × 10−5 | ||
The unit cell (space group P21/c) parameters a, b, c and V decrease continuously with decreasing temperature (see Table 5) in the 2-athdClO4 crystal structure with the values of Δa/a = 1.31%, Δb/b = 0.63%, Δc/c = 0.60% and ΔV/V = 2.72%. Conversely, the cell parameter β increases with the decreasing temperature (see Fig. 9d) with the overall value of Δβ/β = −0.58%. The temperature dependence of the unit cell parameters a, c and β can be approximated by two linear sections with an intersection at ~200 K (see Fig. 9d for an example). The observed differences in the principal linear thermal expansion/contraction coefficients (see Table 4) allow for the division of the studied temperature interval into two parts (phases) – above and below 200 K. The discontinuity of the linear temperature dependency of the unit cell parameter b (see Fig. 9c) was furthermore observed at 170 K.
| Unit cell parameters of 2-athdClO4 | |||||
|---|---|---|---|---|---|
| Temperature (K) | a (Å) | b (Å) | c (Å) | β (°) | V (Å3) |
| 110 | 5.0011(1) | 18.6527(5) | 7.4285(2) | 102.239(1) | 677.21(3) |
| 130 | 5.0073(1) | 18.6640(6) | 7.4320(3) | 102.169(1) | 678.96(4) |
| 150 | 5.0135(2) | 18.6764(6) | 7.4344(3) | 102.134(1) | 680.56(4) |
| 170 | 5.0219(2) | 18.7121(1) | 7.4400(3) | 102.060(2) | 683.71(5) |
| 190 | 5.0282(2) | 18.7071(6) | 7.4430(3) | 102.026(1) | 684.75(4) |
| 210 | 5.0355(2) | 18.7187(6) | 7.4478(3) | 101.955(1) | 686.79(4) |
| 230 | 5.0431(1) | 18.7332(5) | 7.4546(2) | 101.885(1) | 689.16(3) |
| 250 | 5.0512(1) | 18.7464(5) | 7.4608(2) | 101.813(1) | 691.51(3) |
| 270 | 5.0589(1) | 18.7579(5) | 7.4661(2) | 101.733(1) | 693.69(3) |
| 290 | 5.0676(2) | 18.7702(6) | 7.4730(3) | 101.653(1) | 696.18(5) |
The crystal structures of 2-athd2ClO4 were determined at three different temperatures – i.e., 150, 283 and 315 K – due to the temperature limitations of our single-crystal X-ray diffractometer in the higher temperature region and the existence of the thermal effect around 300 K. The only change observed in these three structures is the disorder of the perchlorate anions present at 315 K. The values of the unit cell (space group P21/n) parameters a, β and V increase, whereas the b and c parameters slightly decrease with increasing temperature (Table 6). A very small difference was also observed in the donor⋯acceptor distances of the hydrogen bonds.
| Unit cell parameters of 2-athd2ClO4 | |||||
|---|---|---|---|---|---|
| Temperature (K) | a (Å) | b (Å) | c (Å) | β (°) | V (Å3) |
| 150 | 4.9986(1) | 20.1080(5) | 11.0782(3) | 90.725(3) | 1113.40(5) |
| 283 | 5.1115(2) | 20.0903(6) | 11.1387(5) | 91.663(2) | 1143.37(8) |
| 315 | 5.2439(4) | 19.9290(13) | 11.0435(9) | 92.124(3) | 1153.32(15) |
The values of the interplanar angles between the thiadiazole rings and the plane formed by the NH2 groups (see Table 17S; ESI†) reflect the character of the hydrogen bonds and the complexity of the crystal structures, and therefore, can be used for the evaluation of the observed thermal effects. These values increase with increasing temperature in the case of 2-athdHSO4 (Δφ/φ = 44.52%) and 2-athdClO4 (Δφ/φ = 84.96%) where a sufficient amount of temperature-dependent structural data is available (see Table 20S; ESI†). The slope of the temperature dependence of the interplanar angle for 2-athdHSO4 (see Figure 17aS; ESI†) changes at approximately 200 K, and the dependence also exhibits a discontinuity at 130 K. A similar dependence plot for 2-athdClO4 (see Fig. 17bS; ESI†) can be divided into two parts – above and below the temperature of 190 K.
The temperature-dependent FT Raman spectra of 2-athd2SeO4H2O exhibit only minimal changes during the heating from liquid nitrogen temperature to room temperature. Most of the changes can be observed in the regions of 1280–1200, 920–680 and 455–355 cm−1 (see Fig. 18S; ESI†) at temperatures above the DSC effect (187 K). The splitting of the Raman bands at approximately 1255 and 745 cm−1 is significant. The low temperature FTIR spectra of 2-athd2SeO4H2O are characterized by the narrowing and separation of the vibrational bands, especially in the 1000–600 cm−1 region (see Fig. 19S; ESI†) and the far IR region. Several bands are sensitive to heating the sample to room temperature. The important examples are the low-temperature bands at 1748, 1713, 1507, 1315 and 998 cm−1, which disappeared at temperatures above the DSC effect. The above-mentioned sensitivity of the selected IR bands can also be demonstrated by the temperature dependency of the band position. An example of this behaviour (see Fig. 10a) is the band recorded at 669 cm−1 (room temperature), which can be assigned28 to mixed ν CS, γ rg and δ NH vibrations. It is apparent that the change in the slope of this dependence corresponds well to the temperature of the DSC effect.
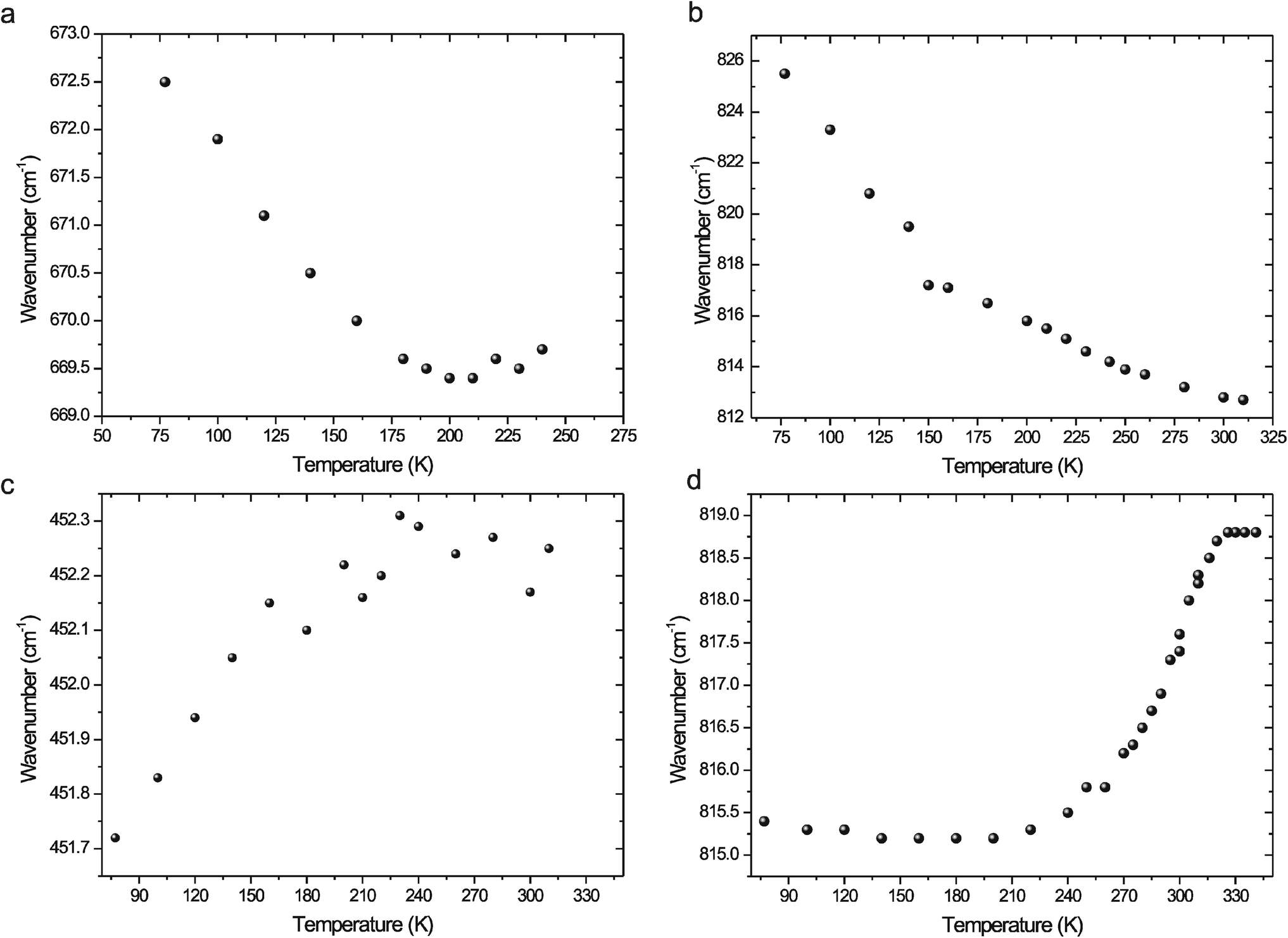 | ||
| Fig. 10 Temperature dependence of the position of selected bands from FTIR spectra: (a) a band at 669 cm−1 of the 2-athd2SeO4H2O spectrum; (b) a band at 813 cm−1 of the 2-athdHSO4 spectrum; (c) a band at 452 cm−1 of the 2-athdClO4 spectrum; (d) a band at 818 cm−1 of the 2-athd2ClO4 spectrum. Mentioned band positions were recorded at room temperature. | ||
Additionally, the FT Raman spectra of 2-athdHSO4 exhibit only a few changes caused by the heating of the sample to room temperature. The separation/splitting of the bands in the 630–550 cm−1 region and the band(s) at approximately 400 cm−1 (asymmetric doublet) is present only in low temperature spectra up to a temperature of 230 K (see Fig. 20S; ESI†), which can be correlated with the endothermic, λ-shaped anomaly recorded in the heating runs in the DSC curves. Similarly, the separation of the bands in the FTIR spectra, especially in the 1500–1400, 1000–750 and 700–500 cm−1 regions (see Fig. 21S – ESI†), is present only below this temperature. Conversely, the temperature dependence of the peak position of the selected bands recorded at 813 cm−1 (300 K) in the FTIR spectra (assigned28 to mixed γ CH and γ OH⋯O vibrations) exhibits a discontinuity at approximately 150 K (see Fig. 10b) – i.e., the temperature matching the reproducible, weak thermal anomaly in the DSC records.
There are no changes in the FT Raman spectra associated with the heating of the 2-athdClO4 sample from liquid nitrogen temperature up to room temperature (see Fig. 22S; ESI†). In addition to the changes observed in the far-IR spectra, most of the recorded changes are located in the mid-IR region (see Fig. 23S – ESI†). Low temperature spectra are characterised by the separation/splitting of bands, especially in the 1200–900 cm−1 and 800–650 cm−1 regions. Such effects are absent at temperatures above 240 K (i.e., temperatures approximately 30 K below the thermal anomaly recorded in the heating run of the DSC measurements). The appearance of a new band at 1513 cm−1 at higher temperatures is significant. Fig. 10c shows the temperature dependence of the position of the band recorded at 452 cm−1 (room temperature), which was assigned28 to the ν2 vibration of the ClO4− anions. This dependence changes at approximately 200 K.
The FT Raman spectra of 2-athd2ClO4 exhibit only a few changes during the heating from room temperature to 340 K (see Fig. 24S; ESI†). The only noticeable changes in the relative intensities of the bands are observed at approximately 1510 and 400 cm−1, which are especially apparent above 300 K – i.e., the temperature corresponding to the thermal anomaly present in the DSC curves. In addition to the changes in the stretching vibrations of the hydrogen-bonded N–H groups (3450–3330 and 3110–2740 cm−1), the FTIR spectra reflect the heating of the sample through intensity changes in the bands in the 1630–1615 cm−1 region (see Fig. 25S; ESI†). Additionally, the bands recorded at ~1100 and 630 cm−1, which were assigned28 to perchlorate ν3 and ν4 vibrations, respectively, exhibit a symmetrising tendency at temperatures above 300 K. This behaviour is in agreement with the disorder of the ClO4− anions present in the crystal structure at higher temperatures. A detailed analysis of the changes in the band positions during the heating of the sample indicated several interesting bands at 1341, 818 and 692 cm−1. The temperature-induced shift of the band at 818 cm−1 assigned28 to δ CH vibrations is present in Fig. 10d. The inflection point of this dependence can be correlated with the temperature of the thermal anomaly found in the DSC records.
If we are trying to summarise the results of the above-mentioned thermal studies, it is obvious that in the case of 2-athd2SeO4H2O the temperature-dependent vibrational spectra are consistent with the DSC measurements and confirm the existence of a weak thermal effect at approximately 190 K, which is associated with minimal changes in the crystal structure.
The results of the low temperature vibrational spectroscopic study and the X-ray structural analysis roughly correlate with the presence of the three types of thermal effects found in the DSC curves of 2-athdHSO4 at 160, 212 and 236 K. These findings can also be confirmed through a detailed analysis of the temperature dependence of the interplanar angles in the cation (see Fig. 17aS; ESI†) and the temperature dependence of the donor⋯acceptor distances in selected O–H⋯O and N–H⋯O hydrogen bonds – i.e., changes in the slope of dependence at ~160 K (see Fig. 11) and the existence of a discontinuity in the 200–250 K region (see Fig. 26S; ESI†).
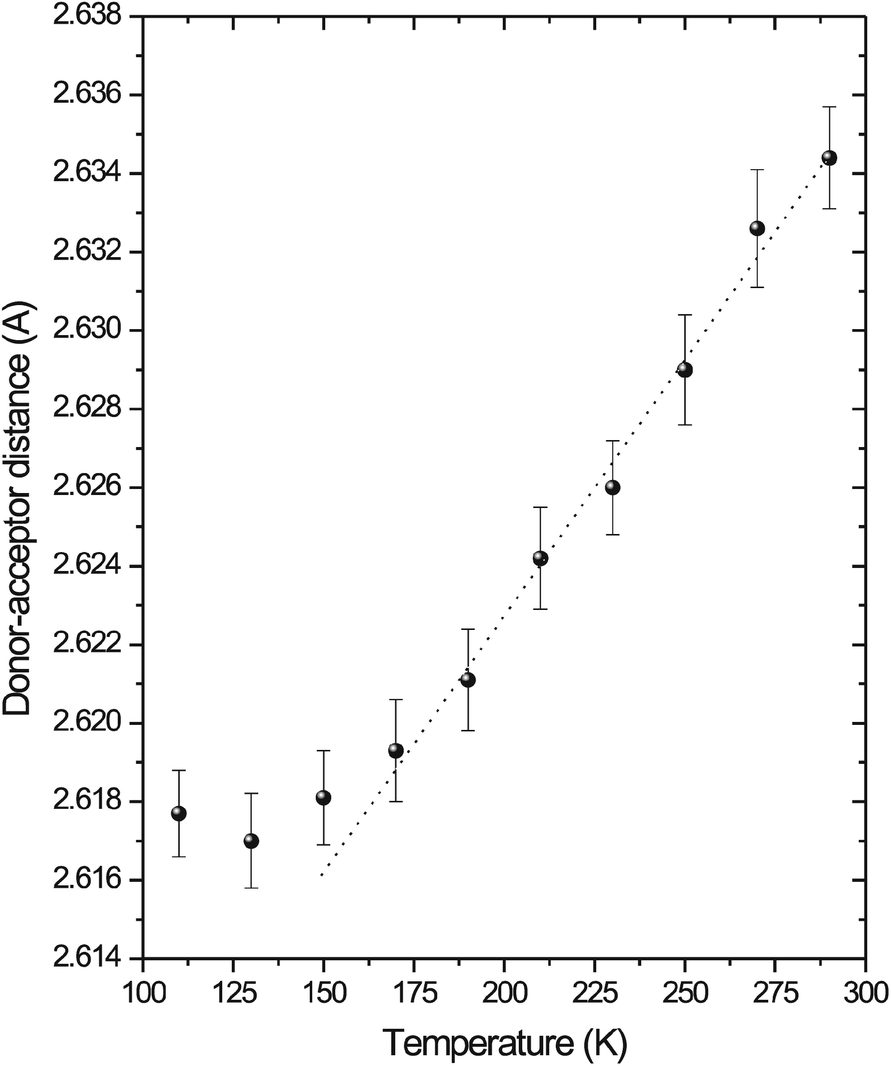 | ||
| Fig. 11 Temperature dependence of the donor⋯acceptor (O4⋯O3c) distance of the O–H⋯O hydrogen bond in 2-athdHSO4. Dashed line represents ideal linear dependence. | ||
In the case of 2-athdClO4, we can consider two “phases” above and below ~200 K (according to the X-ray structural analysis and the vibrational spectroscopic study). This conclusion is also consistent with the temperature distribution of the interplanar angles in the cation (see Figure 17bS; ESI†) and the changes in the slope of the temperature dependence of donor⋯acceptor distances in selected N–H⋯O hydrogen bonds (see Fig. 27S; ESI†). Surprisingly, the corresponding effects were recorded at much higher temperatures in the DSC measurements. The reasons for this discrepancy are unclear, but it could also be influenced by the thermal hysteresis in the DSC results and the different approaches of the measurements; DSC is a dynamic method, whereas X-ray diffraction and vibrational spectroscopic measurements are static (“step by step”) methods.
Second harmonic generation
The calculations (B3LYP/6-311+G(d,p)) of the first hyperpolarisability values for the 2-athd molecule (βtot = 4.47 × 10−30 esu) and the 2-athd(1+) cation (βtot = 2.04 × 10−30 esu – see Table 7) showed that they are very interesting moieties with a total hyperpolarisability comparable with the standard molecule of urea (βtot = 7.77 × 10−31 esu for B3LYP/6-311G).| β xxx | β xxy | β xyy | β yyy | β xxz | β xyz | β yyz | β xzz | β yzz | β zzz | β tot | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Note: (516.84 a.u. = 4.47 × 10−30 esu; 235.89 a.u. = 2.04 × 10−30 esu). | |||||||||||
| 2-athd | −478.91 | −73.11 | −9.42 | −65.75 | 10.08 | 7.20 | −1.65 | 4.71 | −43.33 | −2.04 | 516.84 |
| 2-athd(1+) | −209.03 | −32.10 | −1.80 | −14.66 | 0.00 | 0.00 | 0.00 | −7.63 | −42.23 | −0.01 | 238.89 |
Non-centrosymmetric crystals of 2-athdH2PO4, 2-athd2SeO4H2O and 2-athdHSO4 are quite promising compounds for SHG with relative efficiencies d2-athdH2PO4 = 0.8dKDP, d2-athdSeO4 = 0.8dKDP and d2-athdHSO4 = 0.5dKDP for a 1064 nm excitation laser, which were determined using the standard Krutz–Perry experimental arrangement29 where transmitted radiation is detected. The consecutive measurements in a back-scattering geometry10,12,30 (which generally exhibit lower efficiency values but have higher reproducibility due to improved sample packing) yield relative efficiencies of d2-athdH2PO4 = 0.4dKDP, d2-athdSeO4 = 0.2dKDP and d2-athdHSO4 = 0.1dKDP for an 800 nm excitation laser. These observed differences, which are most likely caused by the different experimental geometry rather than by the spectral dependence of the SHG efficiency, also reflect the fact that the obtained relative efficiencies of powdered samples should be regarded as only approximate and starting values for further NLO characterisation of bulk single crystals.
Conclusions
The preparation of eight novel compounds, 2-athdClH2O, 2-athdClO4, 2-athd2ClO4, 2-athdNO3, 2-athdHSO4, 2-athd2SeO4H2O, 2-athdH2PO4 and 2-athdHPO3, was motivated by their potential use in non-linear optical applications. The majority of the prepared crystals belong to the monoclinic system with non-centrosymmetric space groups Pc (2-athd2SeO4H2O) and Cc (2-athdH2PO4) and centrosymmetric space groups P21/c (2-athdClH2O, 2-athdClO4, 2-athdNO3), P21/a (2-athdHPO3) and P21/n (2-athd2ClO4). Only crystals of 2-athdHSO4 belong to the orthorhombic non-centrosymmetric group P212121.The simplest of all these hydrogen-bonded structures is the layered crystal structure of 2-athdNO3. The more complicated structure of 2-athdClO4 is formed by a 3D network of tetrahedral perchlorates and 2-athd(1+) cations. Structures of 2-athd2ClO4 and 2-athdClH2O contain hydrogen-bonded pairs formed by the 2-athd molecule and the 2-athd(1+) cation or by two 2-athd(1+) cations, which are involved with anions and/or water molecules in 3D networks. Cationic chains were observed in the crystal structure of 2-athd2SeO4H2O. The structures of 2-athdHSO4 and 2-athdH2PO4 are based on anionic chains (2-athdHSO4) or on an anionic 3D network (2-athdH2PO4) incorporating 2-athd(1+) cations. The remaining two crystal structures of 2-athd2SeO4H2O and 2-athd2HPO3 are formed by a network of alternating cations and anions.
Only the combination of DSC, X-ray structural analysis, FT Raman and FTIR spectroscopy enabled a deeper understanding of the thermal behaviour of 2-athd2SeO4H2O, 2-athdHSO4, 2-athdClO4 and 2-athd2ClO4 crystals. The observed effects, which were recorded mostly at low temperatures, are generally associated with minimal changes in the crystal structures and the preservation of crystal symmetry. Therefore, we can consider them interesting examples of isostructural phase transformations.
The results of the theoretical calculations of the first hyperpolarisability values for 2-amino-1,3,4-thiadiazole and its cation, which indicate their potential in the field of nonlinear optics, were confirmed by the measurement of the SHG efficiency of powdered samples of non-centrosymmetric 2-athd2SeO4H2O, 2-athdHSO4 and 2-athdH2PO4 at 1064 and 800 nm. The total SHG efficiency of the prepared crystals is crucially affected by the arrangement of thiadiazolium cations (as carriers of NLO properties) in crystal structures. The arrangement of 2-athd(1+) cations along the 21 screw axes, which are mutually perpendicular in the crystal of 2-athdHSO4 (P212121), leads to lowest SHG efficiency. Higher efficiency was observed in the case of 2-athd2SeO4H2O (Pc), where two independent cations are present in the asymmetric unit and both of them are perpendicularly oriented to the glide plane c. The highest value of SHG efficiency was observed for 2-athdH2PO4 (Cc), where the asymmetric unit contains only one 2-athd(1+) cation, which is also perpendicular to the glide plane c. The crystals of 2-athd2SeO4H2O and 2-athdH2PO4 are the most promising materials with an efficiency of 80% of the KDP standard (1064 nm excitation). All materials are transparent down to 245 nm and thermally stable up to their melting points (at least 378 K).
Acknowledgements
The study is a part of a Long Term Research Plan of the Ministry of Education of the Czech Republic no. MSM0021620857.Notes and references
- R. W. Boyd and G. L. Fischer, Encyclopedia of Materials: Science Technology, Elsevier Science Ltd., 2001 Search PubMed.
- Ch. Bosshard, K. Sutter, P. Pretre, J. Hulliger, M. Florsheimer, P. Kaatz and P. Gunter, Organic nonlinear optical materials (Advances in nonlinear optics series), Gordon and Breach Publishers SA, Amsterdam, 1995, 114 Search PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397 RSC.
- G. R. Desiraju and T. Steiner, The weak hydrogen bond, Oxford University Press, Oxford, 1999 CrossRef CAS; G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Inc., New York, 1997 CrossRef CAS; A. Novak, Struct. Bonding, 1973, 18, 177 CrossRef CAS; G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 CrossRef; L. J. Bellamy and A. J. Owen, Spectrochim. Acta, Part A, 1969, 25, 329 CrossRef.
- C. B. Aakeroy, P. B. Hitchcock and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1992, 553 RSC.
- C. B. Aakeröy, P. B. Hitchcock, B. D. Moyle and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1989, 23, 1856 RSC.
- D. Xue and S. Zhang, J. Phys. Chem. Solids, 1996, 57, 1321 CrossRef CAS; D. Xue and S. Zhang, J. Phys. Chem. A, 1997, 101, 5547 CrossRef.
- M. He, Y. Zhou, R. Liu, J. Dai, Y. Cui and T. Zhang, Dyes Pigm., 2009, 80, 6 CrossRef CAS PubMed.
- I. Matulková, I. Císařová and I. Němec, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o18 Search PubMed.
- I. Matulková, I. Němec, I. Císařová, P. Němec and Z. Mička, J. Mol. Struct., 2007, 834–836, 328 CrossRef PubMed; I. Matulková, I. Němec, K. Teubner, P. Němec and Z. Mička, J. Mol. Struct., 2008, 873, 46 CrossRef PubMed.
- I. Matulková, J. Cihelka, M. Pojarová, K. Fejfarová, M. Dušek, P. Vaněk, J. Kroupa, R. Krupková, J. Fábry and I. Němec, CrystEngComm, 2012, 14, 4625 RSC.
- I. Matulková, I. Císařová, P. Němec, J. Kroupa, P. Vaněk, N. Tesařová and I. Němec, J. Mol. Struct., 2013, 1044, 239 CrossRef PubMed.
- I. Matulková, J. Cihelka, I. Němec, M. Pojarová and M. Dušek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 37, o3410 Search PubMed; I. Matulková, I. Němec, J. Cihelka, M. Pojarová and M. Dušek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o3216 Search PubMed.
- J. Matysiak, Chem. Pharm. Bull., 2006, 54, 988 CrossRef CAS; W. Rzeski, J. Matysiak and M. Kandefer-Szerszen, Bioorg. Med. Chem., 2007, 17, 3201 CrossRef PubMed; Q. Zeng, M. P. Bourbeau, G. E. Wohlhieter, G. Yao, H. Monenschein, J. T. Rider, M. R. Lee, S. Zhang, J. Lofgren, D. Freeman, C. Li, E. Tominey, X. Huang, D. Hoffman, H. Yamane, A. S. Tasker, C. Dominguez, V. N. Viswanadhan, R. Hungate and X. Zhang, Bioorg. Med. Chem. Lett., 2010, 20, 1652 CrossRef PubMed; S. Kanjeekal, A. Chambers, M. Fung, K. Fung and S. Verma, Gynecol. Oncol., 2005, 97, 624 CrossRef PubMed.
- M. A. Hilfiker, N. Wang, X. Hou, Z. Du, M. A. Pullen, M. Nord, R. Nagilla, H. E. Fries, C. W. Wu, A. C. Sulpizio, J.-P. Jaworski, D. Morrow, R. M. Edwards and J. Jin, Bioorg. Med. Chem. Lett., 2009, 19, 4292 CrossRef CAS PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- G. M. Sheldrick, Program for Refinement from Diffraction Data, University of Göttingen, Germany, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Rodrigues-Carvajal and T. Roisnel, FullProf.98 and WinPLOTR, Applications for Diffraction Commission For Powder Diffraction, International Union for Crystallography, Newletter N°20, 1998 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.2, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Frich, R. D. Dennington, T. A. Keith, J. Millam, A. B. Nielsen, A. J. Holder and J. Hiscocks, Gauss View Version 4.1.2., Gaussian, Inc., Wallingford CT, 2006 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- N. Boechat, S. B. Ferreira, C. Glidewell, J. N. Low, J. M. S. Skakle and S. M. S. Wardell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o42 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS; M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef.
- E. E. Bendeif, S. Dahaoui, M. Francois, N. Benali-Cherif and C. Lecomte, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 700 Search PubMed.
- I. Matulková and I. Němec, Vib. Spectrosc. Search PubMed , will be published.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798 CrossRef CAS PubMed.
- I. Matulková, I. Němec, I. Císařová, P. Němec and Z. Mička, J. Mol. Struct., 2008, 886, 103 CrossRef PubMed; I. Matulková, I. Němec, I. Císařová, P. Němec and P. Vaněk, J. Mol. Struct., 2010, 966, 23 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental powder data for 2-athdNO3, 2-athdClH2O, 2-athd2ClO4, 2-athdClO4, 2-athdHPO3, 2-athdH2PO4, 2-athd2SeO4H2O and 2-athdHSO4; selected bond lengths [Å] and angles [°] for 2-athdNO3, 2-athdClH2O, 2-athd2ClO4, 2-athdClO4, 2-athdHPO3, 2-athdH2PO4, 2-athd2SeO4H2O and 2-athdHSO4; comparison of geometric parameters; atom numbering of all title compounds; anionic chains in the crystals of 2-athdClH2O and 2-athdH2PO4; packing scheme of 2-athdClH2O; layers of 2-athdHPO3; DSC curves of 2-athd2SeO4H2O, 2-athdHSO4, 2-athdClO4 and 2-athd2ClO4; temperature dependence of the cell parameters of 2-athdHSO4 and 2-athdClO4; temperature dependent interplanar angle of cations in 2-athdHSO4 and 2-athdClO4; vibrational spectra at different temperatures of 2-athd2SeO4H2O, 2-athdHSO4, 2-athdClO4 and 2-athd2ClO4; temperature dependent donor⋯acceptor distances of hydrogen bonds in 2-athdHSO4 and 2-athdClO4. CCDC 910444–910450, 910853. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42306a |
| This journal is © The Royal Society of Chemistry 2014 |