 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experiences with applications of macromolecular tools in supramolecular crystallography†
Michał
Wierzbicki
a,
Mirosław
Gilski
bc,
Kari
Rissanen
*d,
Mariusz
Jaskólski
*bc and
Agnieszka
Szumna
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
bCenter for Biocrystallographic Research, Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan, Poland. E-mail: mariuszj@amu.edu.pl
cDepartment of Crystallography, Faculty of Chemistry, A. Mickiewicz University, Poznan, Poland
dDepartment of Chemistry, Nanoscience Center, University of Jyvaskyla, P.O. Box 35, FI-40014, Finland. E-mail: kari.t.rissanen@jyu.fi
First published on 13th January 2014
Abstract
Supramolecular structures, with ever increasing size ranging from a few up to tens of nanometres, represent an intermediate stage between small molecules and biological macromolecules. Many crystal structures of these large supramolecular assemblies have been solved using dual space algorithms. However, supramolecular assemblies with a capsular shape present a particular challenge for crystallography, especially when they are chiral and composed of only light atoms. In this paper, we show that the application of “routine” macromolecular tools may be of great help in solving the crystal structures of supramolecular assemblies that are otherwise refractory to the routine methods of small molecule crystallography. Specifically, we have applied the method of molecular replacement as implemented in PHASER in order to solve the crystal structure of a chiral organic capsule, which could not be determined using direct or dual space methods. By utilizing various models consisting of well-defined supramolecular “bricks” or modelled structures, we show how model size (fraction of the asymmetric unit) and quality (root mean square deviation from target) influence the success rate of medium sized non-protein structures. The results indicate that supramolecular structures, that are still “small molecules” for macromolecular standards, can be successfully solved using even very small models, down to 25% by weight of the contents of the asymmetric unit.
Introduction
Although the underlying principles of macromolecular (also referred to as protein) crystallography and small-molecule crystallography (here concerned mainly with organic molecules) are essentially the same, the experimental and computational approaches in these two areas are quite distinct, partly as a result of tradition, and partly because of real idiosyncrasies of these domains. This is visible, for instance, in the usually simple crystallization methods in small-molecule crystallography and the highly developed, usually high-throughput, miniaturized and robotized macromolecular crystallization techniques, and in the fact that most organic crystal structures are being solved automatically and routinely by direct methods, while this approach is inapplicable for typical-size macromolecular structures. A dramatic divide concerns the achievable resolution, which with small molecules is almost always very high (e.g. about 0.8 Å even when limited by the wavelength of Cu Kα radiation), and in protein crystallography is still rare even at the nominal atomic resolution of 1.2 Å defined by Sheldrick,1 as illustrated by the constant level of ~2% of such structures2 in the Protein Data Bank.3 The latter aspect is responsible, for example, for the fact that while protein crystallographers always build their models on electron density maps, such maps almost never meet the eye of small-molecule crystallographers, who can work quite comfortably with atom/peak lists generated by automatic interpretation of such maps. There are, however, also evident lines of convergence. For example, the loop method introduced for mounting protein crystals for cryogenic experiments4 is gaining popularity in small-molecule crystallography, and the two communities use cryogenic temperatures for routine data collections (although the reasons in the two cases may be somewhat different). Also, the high-resolution barrier is gradually becoming a historical one, as record-breaking ultrahigh-resolution structures of proteins (0.48 Å)5 and nucleic acids (0.55 Å)6 can now also be found in the PDB. A very encouraging example of convergence is provided by the highly popular SHELX system of crystallographic programs,7 originally developed for small-molecules and later very successfully converted by its author to a versatile system, now also widely used in macromolecular crystallography. In general, however, the computational tools are quite different in the two communities, the small-molecule programs usually being incapable of handling the huge macromolecular cases and the macromolecular programs being often hard-wired for the structural specifics of biopolymers.In this work, we demonstrate that with very little additional effort, the powerful computational tools of protein crystallography can become extremely useful in small molecule crystallography as well, especially when the “small molecules” are not so small at all, as is the case of self-assembling supramolecular systems. We show that application of “routine” macromolecular tools may greatly help with solving crystal structures of supramolecular assemblies that cannot be solved using “routine” small molecule crystallography methods.
Results and discussion
Experimental problems with crystal structure determination of capsular assemblies
For the last few decades, most small-molecule structures have been routinely solved by direct methods. Direct methods are a subclass of ab initio methods, i.e. methods of crystal structure determination that do not require prior knowledge of any atomic positions. Direct methods algorithms, as implemented in the programs SHELXS7 and SIR,8 usually are not effective with more than ~200 non-hydrogen atoms, although there are cases of spectacular success even with much larger structures.9–11 The more recently developed methods based on dual space algorithms, as implemented in, for example, the programs Shake-and-Bake,12 SHELXD7 or SUPERFLIP,13 have significantly pushed the boundaries of ab initio crystallographic methods. All ab initio methods rely on the atomicity constraint, which is implemented by requiring the electron density to be concentrated at randomly distributed, resolved, and equal-atom positions. The mathematical solution requires atomic-resolution data, which is often difficult to achieve for macromolecular structures and also for some supramolecular structures. However, when the atomicity condition is met (i.e. accurate diffraction data have been measured to a resolution of 1.2 Å or better), the dual space methods have proven capable of solving complete structures containing as many as 2000 independent non-H atoms.Supramolecular structures (artificially constructed non-covalent assemblies), with their increasingly larger size ranging from a few nanometers up to tens of nanometers, are located at the interface between small molecules and macromolecules. With the spectacular advances in data quality (mostly owing to synchrotron radiation sources)14 and development of dual space algorithms, many crystal structures of large supramolecular assemblies are now solved using ab initio methods.15 However, the special case of assemblies with a capsular shape presents a particular challenge for crystallography, particularly when they are made of light atoms and are chiral, like those that have been recently the subject of interest of our group.16–19 There are good structural reasons for that. Capsular assemblies, by definition, contain vaults or cavities that are often filled with highly disordered solvent. Since the capsules resemble large spherical objects, their inter-capsular voids are also quite large and often also filled with disordered solvent. Therefore, the volume ratio between the well-defined capsule framework and the poorly defined regions can be quite low. Organic capsules are usually crystallized from organic solvents, which makes the crystals fragile and prone to fast decomposition due to solvent volatility. Such crystals often diffract poorly and only low resolution data can be obtained. Further problems that are particularly relevant to capsular assemblies are related to their high symmetry. Symmetry may be of great help if the capsule molecular symmetry coincides with crystallographic symmetry. However, in many cases, the molecular symmetry does not overlap or only approximately overlaps with crystal lattice symmetry. As a result of this symmetry mismatch, the crystals are very prone to twinning, and this effect is aggravated by the often weak inter-capsular interactions. Therefore, space group determination may be problematic because of twinning and pseudosymmetry.
Solution of the problem – molecular replacement
Despite the above difficulties, one great structural advantage of supramolecular capsules is that they are often built from well-defined molecular “bricks”. Out of the arsenal of several known “bricks”, chemists have made an enormous number of covalent derivatives and sophisticated multicomponent assemblies. For example, there are more than 2500 structures containing calix[4]arene skeleton, ~960 resorcin[4]arenes, and ~690 various cucurbiturils (CSD, version 5.34).20 The advantage of these “bricks” consists in the fact that their 3D structures are well known and in most cases not susceptible to conformational changes (at least not considerable). Because of that, supramolecular crystallography can benefit from building crystal structures from known molecular fragments, i.e. can exploit the methods of molecular replacement.Molecular replacement (MR) involves the rigid-body placement of a search model in the asymmetric unit of the target crystal, with the aim of finding the best match between the search model and the target structure. Computer programs for MR have been around for several decades. The success of the method depends predominantly on two factors: the fraction of the asymmetric unit for which there is a suitable model, and the RMS deviation (after optimal superposition) between the model and target structures. Although the availability of a good model is a prerequisite for MR, the quality of the target functions and the search strategy are also important for success. Traditionally, molecular replacement has been based on the properties of the Patterson function. The factors that can complicate the problem are high symmetry, tight packing and/or multiple search components in the asymmetric unit. Large numbers of components in the asymmetric unit are particularly problematic for traditional MR algorithms, where each component of the asymmetric unit is found independently and therefore the fraction of the total scattering contributed by each component is low. As an alternative to Patterson-based MR, a maximum-likelihood (ML) MR approach is now playing an increasingly important role, particularly for difficult molecular replacement problems.21–23 The ML algorithm is implemented, for example, in the program PHASER.24 The method significantly improves the success rate in cases where there are multiple search components in the asymmetric unit because it has more discriminating (maximum-likelihood) rotation and translation functions than other methods, and these functions also utilize the information about the orientation and translation of a given component to increase the signal-to-noise ratio of both the rotation and translation search for other components. A recent PHASER version also finds pseudo-translational NCS (non-crystallographic symmetry) and corrects the data for intensity variations using likelihood methods, yielding molecular replacement solutions with even higher signal-to-noise level.23 The ML MR functions implemented in PHASER have allowed many previously intractable macromolecular MR problems to be solved.23,25–27
Working example – chiral organic capsule with polar interior
The example presented to illustrate our approach is a chiral organic capsule, 1, non-covalently assembled from two chiral hemispheres (Fig. 1).19 The assembly motif consists of a system of ionic hydrogen bonds (salt bridges) between carboxylic and amine groups. The capsule has a reversed polarity (in analogy to reverse micelles), with polar groups gathered inside and a hydrophobic outer shell. The hemispheres consist of resorcin[4]arene scaffolds decorated with four L-alanine arms. Although the amino acid arms are highly flexible, solution studies have indicated that the association motif for 1 is similar to that reported previously for L-phenylalanine analogue 2.16 Therefore, we expected that the present structure would have a capsular shape with a cavity volume of ~310 Å3, capable of binding small polar molecules.17,18 Considering the size of the unit cell and its contents, the structure presents a medium-sized crystallographic problem according to current standards (Table 1). The quality of the dataset, which extends to 0.90 Å resolution, is quite high, as indicated by Rint = 0.044 for orthorhombic lattice. Statistics of the systematic absences indicated the P212121 space group. However, our previous experience suggests that some of the reflection intensities might be artificially low due to the high symmetry of the assemblies. Therefore, in the subsequent MR calculations, we tested all primitive space groups in the 222 class by allowing all possible combinations of twofold and twofold screw axes (2 and 21).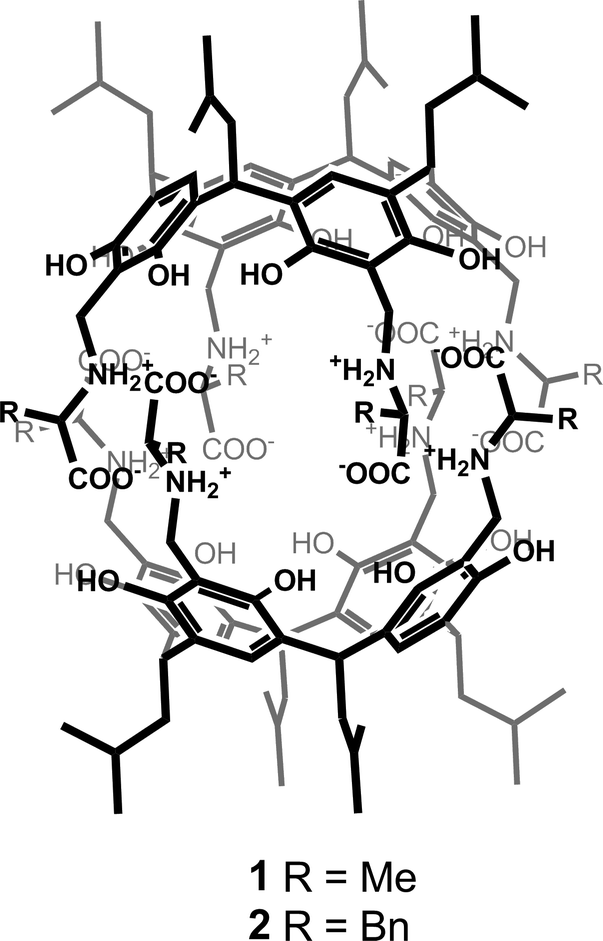 | ||
| Fig. 1 Chemical structures of the chiral capsules with reversed polarity. | ||
| Crystal data | |
|---|---|
| Moiety formula | 1 (acetone)9.1·(MeCN)0.7·H2O |
| Empirical formula | (C60H84N4O16)2·(C3H6O)9.1·(C2H3N)0.7·H2O |
| Formula weight | 2833.46 |
| Temperature (K) | 150 |
| Wavelength (Å) | 1.54178 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell dimensions a/b/c (Å) | 15.7322(4) |
| 24.0717(7) | |
| 41.3778(12) | |
| Unit cell volume (Å3) | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 669.8(8) 669.8(8) |
| Z | 4 |
| Calculated density (g cm−3) | 1.191 |
| Absorption coefficient (mm−1) | 0.710 |
| F(000) | 6066 |
| θ range for data collection (°) | 58.99–3.00 |
| Index ranges | −11 < h < 17 |
| −26 < k < 24 | |
| −44 < l < 45 | |
| Reflections collected | 33459 |
| Independent reflections (Friedel opposites not merged/merged) | 20458 (Rint = 0.0462) |
| 11872 (Rint = 0.0491) |
|
| Completeness to θmax | 0.967 |
| Refinement statistics (with disordered solvent molecules) | |
|---|---|
| Final R indices [>2σ(I)] | 0.0995 |
| R indices [all data] | 0.1037 |
| Goodness-of-fit | 1.160 |
| Extinction coefficient | 0.00212(10) |
| Largest diff. peak and hole (e Å−3) | 0.471/−0.396 |
| Refinement statistics (with solvent masking procedure) | |
|---|---|
| Final R indices [>2σ(I)] | 0.0757 |
| R indices [all data] | 0.0815 |
| Goodness-of-fit | 1.055 |
| Extinction coefficient | — |
| Largest diff. peak and hole (e Å−3) | 0.463/−0.312 |
The dataset for the crystal of 1 seemed suitable for the ab initio structure solution methods owing to a quite high diffraction quality. We therefore first attempted to solve the structure by ab initio methods using virtually all routine procedures available in SHELXS, SIR and SUPERFLIP. The attempts were carried out independently in laboratories in Poland (Szumna) and in Finland (Rissanen). In those runs, for example SHELXS, produced many trial solutions with very similar figures of merit, none above a conclusive threshold. We also tested SUPERFLIP and SHELXD. In the multi-run mode, SUPERFLIP generated many potential solutions that met the default criteria for convergence. However, none of them had any expected features of the capsule structure.
The previously reported structure of a chiral capsule with L-phenylalanine arms (2) was solved using DIRDIF.28 DIRDIF is a computer program that uses a traditional Patterson-based version of molecular replacement in combination with direct methods in an implementation suitable for small molecule problems. In the past, it has proven to be very successful in our hands for solving many problematic structures, often using twinned data.16,29 However, in the present case, multiple trials using various models (Fig. 2) in all tested space groups have failed to find a solution.
 | ||
| Fig. 2 Chemical structures of models that were tested for solving the structure of 1 using molecular replacement methods. RMSD value and fraction of the final structure (by weight) for each model are given below. The comment indicates if the model resulted in solved structure using PHASER program with default parameters. | ||
As most of the “standard” small-molecule crystallographic tools failed to give a solution, we turned our attention to macromolecular software. The advantages of using maximum-likelihood MR as implemented in PHASER include searching for many independent fragments simultaneously and checking of all chiral space groups within a given crystal class and Bravais lattice. As the initial model, we used the core of capsule 2 (model A1, Fig. 2). With model A1, the structure was easily solved using a standard PHASER procedure (without modification of any of the default parameters) and the program returned the solution in the P212121 space group. The structure was subsequently refined using SHELXH to the final R1 = 0.0995 with modelled solvent disorder or to R1 = 0.0757 with solvent masking procedure in OLEX2.30 The asymmetric unit is composed of dimer 1 with one water and 9.1 acetone molecules and 0.7 of an acetonitrile molecule (Fig. 3). Two acetone molecules and one water molecule are found inside the cavity; the remaining acetone molecules are located between the capsules and mostly disordered. Interestingly, inspection of the contour electron density maps, as it is common for macromolecules, allowed for the unequivocal location of some acetone molecules (Fig. 4). These solvent molecules with partial occupancies are not clearly visible using electron density peaks, typically generated during small molecule refinement. The final structure of capsular dimer 1 is found to deviate significantly from the “model”, at least more than expected (Fig. 3b). The differences involve different conformations of the amino acid arms. In particular, one of the L-alanine arms has a conformation, with the side chain located inside the cavity, that has not been observed in any of the previous structures, either in the solid state or in solution. Additionally, the relative position of the resorcin[4]arene scaffolds is significantly different. In the structure of 2 (used as a model), the dihedral angle between the scaffolds is 44°. In the present structure of 1, this angle is only 29°. As a result of those differences, the RMSD value between the corresponding parts of the structures of capsules 1 and 2 is as high as 1.082 Å (Fig. 3b).
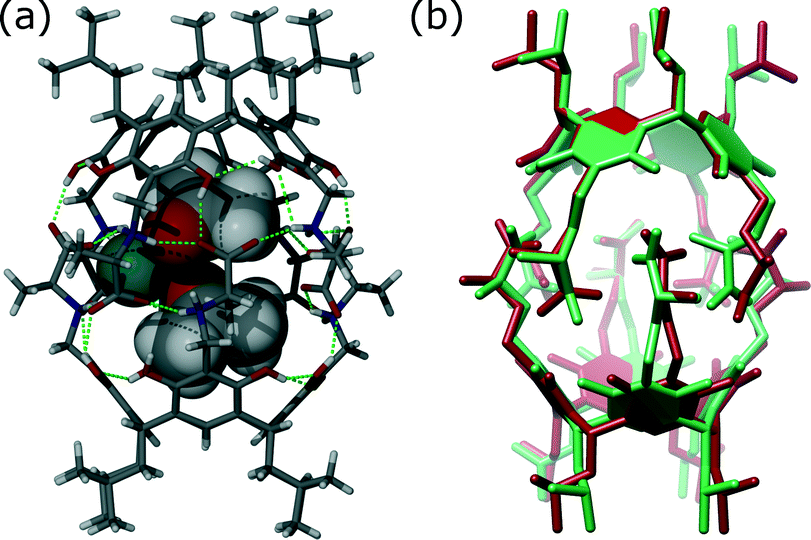 | ||
| Fig. 3 (a) Structure of 1 (sticks) with two molecules of acetone (van der Waals spheres) and one water molecule (aquamarine) trapped inside the cavity; (b) superposition of the corresponding parts of the structures of 1 (green) and 2 (red). | ||
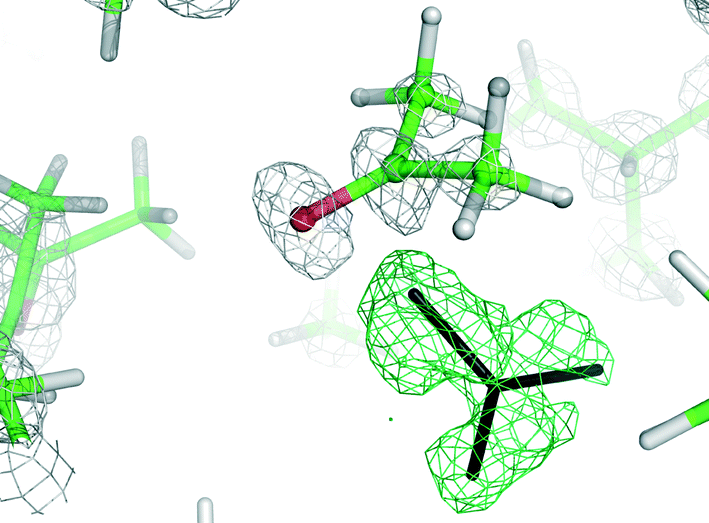 | ||
| Fig. 4 A section of 2Fo − Fc (grey) and difference Fo − Fc (green) electron density maps contoured at the 1.5 and 3.0σ level, respectively. The difference map was used for the identification of an acetone molecule (black sticks). | ||
The A1 model that was used for MR in the initial PHASER attempt accounted for quite a substantial fraction of the unknown structure (60% by weight). Its creation was possible due to the availability of structural data for 2. However, the model is not very accurate in small-molecule standards (RMSD 0.912 Å, Fig. 5a). One can expect that a model of similar quality could also be obtained through molecular modelling. To check this, we constructed a model “from scratch” using the qualitative information on the interaction mode between the monomers and the approximate symmetry from NMR experiments. The fragment was optimized using molecular mechanics (with various force fields). The modelling afforded fragment A2 with visually different positions of the arms and different geometry of the resorcin[4]arene scaffold (Fig. 5b, RMSD 0.994 Å). The modelled fragment A2 also produced a correct solution of the crystal structure in PHASER. This example indicates that molecular modelling with some hints from NMR models can also be of great help in solving crystal structures by molecular replacement.
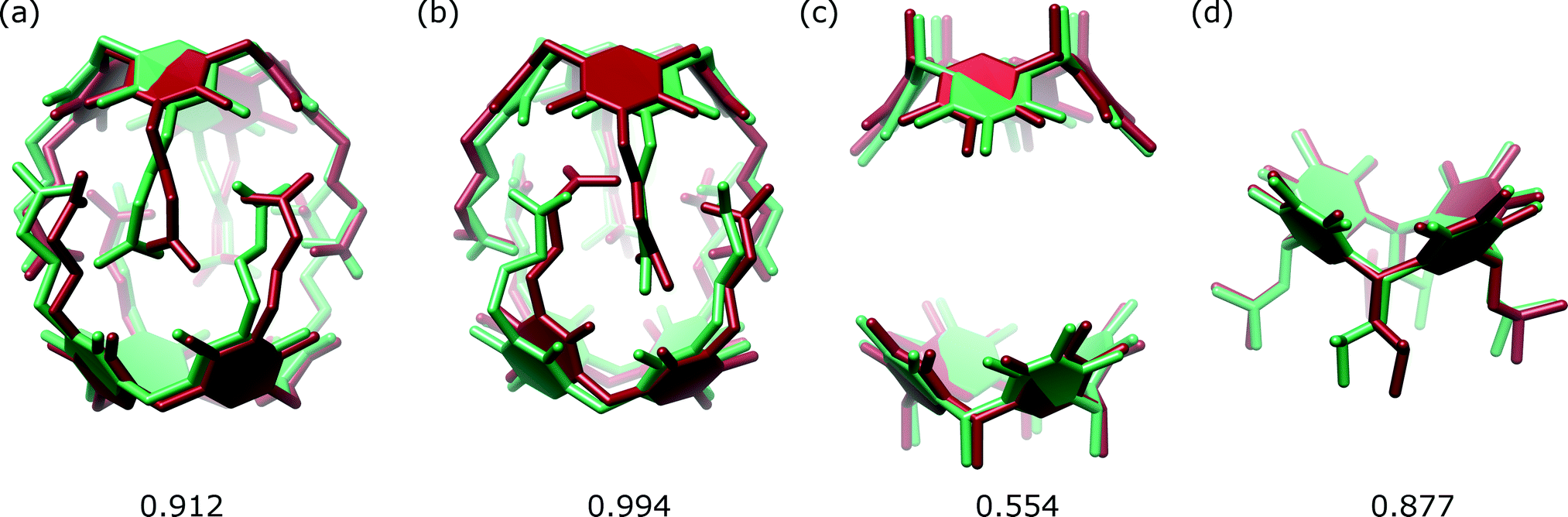 | ||
| Fig. 5 Superposition of the corresponding sections of the structure of 1 (green) and models (red): (a) A1; (b) A2; (c) C and (d) E. The RMSD value for each case is given below in Å. | ||
We also tested whether smaller fragments could be used for solving the crystal structure. We gradually reduced the size of the model down to the most characteristic “brick” of the present capsule, consisting of just the resorcin[4]arene skeleton (Fig. 2). The results show that with the use of default procedures in PHASER, the success rate is not a simple function of model size. Application of the two rigid resorcin[4]arene scaffolds at the correct distance but with wrong relative rotation (model C, model size 40% by weight, RMSD 0.554 Å) still allowed for a successful solution of the structure. Also, a model consisting of only a single resorcin[4]arene skeleton with flexible lower-rim alkyl chains (model E, 25%, RMSD 0.877 Å) allowed us to solve the structure. In this case, even though the location of two copies was requested, PHASER found only one of them (or at least only one was visible after the peak-search interpretation of the electron density map). Thus, initially, only a small part of the structure was available in this case. However, subsequent step-by-step expansion of the model led to successful completion and refinement of the whole structure. With the use of the smallest rigid building block, i.e. the resorcin[4]arene skeleton itself (model F, 20%, RMSD 0.122 Å), the structure cannot be solved by the default runs of PHASER.
An interesting discontinuity in the dependence between model size and the chance of success was observed for models B and D (Fig. 2). Even though both the smaller and larger models gave the correct solution, the medium-sized models did not, although they were not worse in terms of their RMSD values. The reasons for that can be traced down to the packing criterion that is routinely checked by the software. By default for protein structures, the program discards solutions that have too many clashes of Cα atoms (more than 5). For supramolecular structures, it is hard to predict what should be classified as Cα atoms and tighter packing of the subunits is possible. Therefore, in subsequent runs, we allowed PHASER to accept solutions without checking the packing criterion. The calculation time in this case was much longer (ca. five times), but we could obtain the correct solutions for models that were previously unsuccessful (B and D).
Conclusions
In this work we have shown how the powerful computational tools of protein crystallography can be successfully applied to crystal structures of supramolecular assemblies that may not be easily amenable to approaches typical for small-molecule crystallography. Of particular interest are the powerful molecular replacement methods, as many supramolecular structures are assemblies of known fragments. Among those methods, the maximum-likelihood-based MR algorithms with the possibility to simultaneously search for multiple fragments, as implemented in PHASER, seem to be particularly suitable. In many cases, the default parameters used by the MR software can be successfully applied for supramolecular structure solution. However, one has to be aware of the inherent differences between the supramolecular and protein structures, which may require a deviation from the protein-specific default setting. They are mainly related to the substantially different packing characteristics. Although supramolecular structures are quite large by the standards of small molecule crystallography, they are still rather small for typical protein crystallography. A benefit of this is that even very small models can lead to successful structure solution. In the present paper, we have shown that models that are composed of only 25% of the total weight of the asymmetric unit can still yield an appropriate solution of the crystal structure.As the size and complexity of supramolecular structures are constantly growing, one can predict that the number of examples emerging at the interface between small molecules and macromolecular crystallography will also be growing. The present experiences foretell a great promise for the application of macromolecular methodology in supramolecular crystallography and highlight the unity of these two poles of structural crystallography.
Experimental
Materials
Compound 1 was synthesized as previously reported.19 A 10 mg sample of 1 was crystallized from acetone. The crystals were quite large but decomposed within seconds after removal from the mother liquid. The crystals were transferred as soon as possible into a loop containing perfluorinated oil and frozen at 150 K. X-Ray diffraction data were measured on a Bruker Kappa Apex II diffractometer at Polish Academy of Sciences (Warsaw, Poland) equipped with a sealed-tube Cu Kα source, a APEX2 detector and a low-temperature device. APEX2 software was used for the data measurement for the processing. Crystal mosaicity 0.78°. Data were integrated in an orthorhombic P crystal system with LS profile fitting enabled (using default settings). The data were corrected for Lp and absorption (estimated minimum and maximum transmission: 0.7864 and 1.0000).The protocol of structure solution using PHASER
A reflection file obtained in the data reduction process was converted from SHELX hkl to mtz format using the F2MTZ routine of the CCP4 suite. The CTRUNCATE procedure was used to convert the intensities to structure factors, and the scattering power was calculated based on the atom count in the asymmetric unit (excluding any possible solvent, the content of which was not known at that stage). The obtained mtz file was then edited using the SFTOOLS module to input the correct wavelength. The models for molecular replacement (in PDB format) were prepared using X-Seed,31 starting from a previously refined structure of 2. The PHASER program ver. 2.51 from the CCP4 package was used to solve the structure, taking into account all the possible primitive space groups within the given point group symmetry (222 in this case). The RMSD between each model and the target structure was set to 1 Å. The resulting PDB file, containing the oriented and translated atomic model, and mtz file, containing the original diffraction data plus the model-derived structure-factor information and Fourier map coefficients, were then inspected directly in the COOT program, a molecular-graphics application for model building and validation.32 Visualization of the electron density maps calculated on the basis of these data allows one to build and validate the structural model. The peak coordinates located in a peak-search procedure were written in the atomic coordinate PDB file that was subsequently exported to a SHELX res file format using Mercury.33Refinement
The structure was refined with SHELXH using the X-Seed interface.31 All ordered non-hydrogen atoms were refined with anisotropic thermal parameters (ADP). All H atoms were positioned geometrically. The –CH3 hydrogen atoms were staggered with respect to the shortest other bond to the atom to which the –CH3 is attached; water hydrogen atoms were not located. Geometrical restraints were applied for the disordered fragments (FLAT, DFIX, DANG, and SAME). The disorder is mainly observed in the peripheral part of the molecule (alkyl chains) and for the intracapsular acetone molecules. Most of the disordered atoms were located in dual positions. With all solvent molecules located from the electron density peaks, we obtained R1 = 0.0995 (Table 1), CCDC 971032.Inspection of ADP parameters for intracapsular solvent molecules and peripheral alkyl chains indicates that some additional disorder is still possible. However, numerous attempts to model this residual disorder with alternative occupancies did not yield stereochemically reasonable results. As an alternative to the previous refinement, we have applied a solvent masking procedure as implemented in OLEX2.30 We have only left those acetone molecules having full occupancy (two intracapsular and three intercapsular molecules, no restrains) and masked the remaining solvent molecules (disordered). It resulted in a final R1 = 0.0757, CCDC 971033.
Acknowledgements
We acknowledge the financial support of the Polish National Science Centre (AS grant N N204 187839) and the Academy of Finland (KR, grant no. 265328 and 263256). The interdisciplinary cooperation was supported by the Polish Science Foundation (Mentoring programme, 59/U/SKILLS/2013). We would also like to thank Dr. Oleksandr Shkurenko for independent attempts to solve the structure by direct and dual space methods and Dr. Sarah Perry for worthwhile discussions and valuable suggestions.Notes and references
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- M. Jaskolski, in High resolution macromolecular crystallography. In: Advancing Methods for Biomolecular Crystallography, ed. R. Read, A. G. Urzhumtsev and V. Y. Lunin, Springer, 2013 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–42 CrossRef CAS PubMed.
- T. Y. Teng, J. Appl. Crystallogr., 1990, 23, 387 CrossRef CAS.
- A. Schmidt, M. Teeter, E. Weckert and V. S. Lamzin, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 424 CAS.
- K. Brzezinski, A. Brzuszkiewicz, M. Dauter, M. Kubicki, M. Jaskolski and Z. Dauter, Nucleic Acids Res., 2011, 39, 6238 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357 CrossRef CAS.
- K. Mandal, B. L. Pentelute, V. Tereshko, V. Thammavongsa, O. Schneewind, A. A. Kossiakoff and S. B. H. Kent, Protein Sci., 2009, 18, 1146 CrossRef CAS PubMed.
- K. Mandal, B. L. Pentelute, V. Tereshko, A. A. Kossiakoff and S. B. H. Kent, J. Am. Chem. Soc., 2009, 131, 1362 CrossRef CAS PubMed.
- J.-P. Collin, F. Durola, J. Frey, V. Heitz, F. Reviriego, J.-P. Sauvage, Y. Trolez and K. Rissanen, J. Am. Chem. Soc., 2010, 132, 6840 CrossRef CAS PubMed.
- J. Rappleye, M. Innus, C. M. Weeks and R. Miller, J. Appl. Crystallogr., 2002, 35, 374 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- E. M. H. Duke and L. N. Johnson, Proc. R. Soc. London, Ser. A, 2010, 466, 3421 CrossRef CAS.
- M. Kudo, V. Maurizot, B. Kauffmann, A. Tanatani and I. Huc, J. Am. Chem. Soc., 2013, 135, 9628 CrossRef CAS PubMed; Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grelard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2010, 132, 7858 CrossRef PubMed; D. Sanchez-Garcia, B. Kauffmann, T. Kawanami, H. Ihara, M. Takafuji, M. H. Delville and I. Huc, J. Am. Chem. Soc., 2009, 131, 8642 CrossRef PubMed; E. Berni, B. Kauffmann, C. Y. Bao, J. Lefeuvre, D. M. Bassani and I. Huc, Chem.–Eur. J., 2007, 13, 8463 CrossRef PubMed; C. Y. Bao, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Chem.–Eur. J., 2009, 15, 11530 CrossRef PubMed; Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, S. Matsunaga, K. Takada, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef PubMed; P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 1461 CrossRef PubMed; P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 1697 CrossRef PubMed; J.-P. Collin, S. Durot, M. Keller, J.-P. Sauvage, Y. Trolez, M. Cetina and K. Rissanen, Chem.–Eur. J., 2011, 17, 947 CrossRef PubMed; J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. T. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15 CrossRef PubMed; W. Jiang, K. Nowosinski, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen, N. Löw and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 1860 CrossRef PubMed; K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 2, 1235 RSC.
- B. Kuberski and A. Szumna, Chem. Commun., 2009, 1959 RSC.
- A. Szumna, Chem. Commun., 2009, 4191 RSC.
- A. Szumna, Chem.–Eur. J., 2009, 15, 12381 CrossRef CAS PubMed.
- M. Wierzbicki and A. Szumna, Chem. Commun., 2013, 49, 3860 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- L. C. Storoni, A. J. McCoy and R. J. Read, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 432 CrossRef PubMed.
- A. J. McCoy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 32 CrossRef CAS PubMed.
- J. Sliwiak, M. Jaskolski, Z. Dauter, A. J. McCoy and R. J. Read, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 471 CrossRef .
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658 CrossRef CAS PubMed.
- M. Jaskolski, M. Li, G. Laco, A. Gustchina and A. Wlodawer, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 208 CrossRef PubMed.
- L. C. James, P. C. Jones, A. J. McCoy, G. A. Tennent, M. B. Pepys, K. Famm and G. Winter, J. Mol. Biol., 2007, 367, 603 CrossRef CAS PubMed.
- D. D. Rodríguez, C. Grosse, S. Himmel, C. González, I. M. de Ilarduya, S. Becker, G. M. Sheldrick and I. Usón, Nat. Methods, 2009, 6, 651 CrossRef PubMed.
- P. T. Beurskens, G. Beurskens, R. de Gelder, S. Garcia-Granda, R. O. Gould and J. M. M. Smits, The DIRDIF2008 program system, Crystallography Laboratory, 2008, University of Nijmegen, The Netherlands Search PubMed.
- B. Kuberski, M. Pecul and A. Szumna, Eur. J. Org. Chem., 2008, 3069 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS PubMed.
- F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef.
Footnote |
| † CCDC 971032 and 971033. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42288g |
| This journal is © The Royal Society of Chemistry 2014 |