DOI:
10.1039/C3CE42159G
(Paper)
CrystEngComm, 2014,
16, 4706-4714
Tautomeric preference in polymorphs and pseudopolymorphs of succinylsulfathiazole: fast evaporation screening and thermal studies†
Received
24th October 2013
, Accepted 7th December 2013
First published on 11th December 2013
Abstract
Succinylsulfathiazole (SST) is known to exist in seven different crystalline forms, namely two unsolvated polymorphic forms (SST-I, SST-II), a monohydrate (SST/H2O), dihydrate (SST/2H2O) and solvates of butanol (SST/But), pentanol (SST/Pnt) and dioxane (SST/Diox). Most of these forms have been characterised only by IR and PXRD, while the single crystal structures have been determined for SST/But, SST/Pnt, SST/H2O and SST/Diox solvates. Previous studies also noted a lack of reproducibility in preparation of these different forms. Here, we employed a fast evaporation (FE) crystallization method and identified two new solvates from acetone (SST/AcMe) and tetrahydrofuran (SST/THF), as well as determined the single crystal structures for SST-I, SST-II, SST/AcMe and SST/THF. This revealed that SST exclusively adopts the imidine tautomeric form in all its solid form structures, but never the amidine form. The succinyl group of SST shows a conformational flexibility and adopts either anti- or syn-geometry to facilitate hydrogen bonding in the different structures. The study also allowed us to rationalize the hydrogen bonding preferences of various functional groups in all the forms. Notably the neat grinding and liquid assisted grinding methods resulted in only SST/H2O from various solvents, while the FE method produced polymorphs or pseudopolymorphs from different solvents.
Succinylsulfathiazole (SST), also known as sulfasuxidine, is a sulfonamide drug and classified as an ultra long acting drug. According to BCS1 it belongs to either the class II or class IV drug molecules.2SST is used for the prevention and treatment of gastrointestinal infections. This drug has solubility problems and shows unfavourable absorption.3,4 According to Moustafa et al.,5 aqueous suspensions of succinylsulfathiazole exhibit physical instability which causes caking, formation of cement-like precipitates and difficult resuspendability. Hence, SST has been a topic of interest to solid-state pharmaceutical chemists since the 1940s and many attempts have been made to identify its alternative solid forms with superior properties. SST is known to exist in several solid forms, I–VI, as named in previous studies, along with an amorphous form.5 Among them, reportedly, forms I and IV are true polymorphs, forms II and III contain two and one moles of water, respectively, form V contains one mole of butanol and form VI contains one mole of pentanol.5a In due course the single crystal structures of the monohydrate and solvates, butanol, pentanol along with a new form 1,4-dioxan were reported,6 while the rest have been characterized only by IR and powder XRD. The lack of reproducibility in the preparation and confusion over the solvent ratios, as noted in some previous studies, makes the structural determination of these forms significant.5 As the naming of the forms (forms I–VI) in previous studies was also somewhat inconsistent, here we rename them as SST/H2O (monohydrate), SST/2H2O (dihydrate), SST/AcMe (acetone), SST/THF (tetrahydrofuran), SST/But (butanol), SST/Pnt (pentanol), SST/Diox (dioxane) and the true polymorphs of SST, SST-I and SST-II.
This study has also allowed us to examine the tautomerism and hydrogen bonding preferences in the SST solid forms and their selective preparation by using the fast evaporation (FE) method, which we have exploited recently in the context of polymorphs and co-crystal screening.7 The solid forms of succinylsulfathiazole have been characterized by powder and single crystal X-ray diffraction techniques, FTIR, differential scanning calorimetry and thermogravimetric analysis.
1. Experimental section
Materials
Succinylsulfathiazole drug (monohydrate form) was purchased from Sigma-Aldrich. Commercially available solvents were used as received without further purification.
Fast evaporation method
A sufficient amount of SST was dissolved in various boiling solvents, taken in separate conical flasks. The solutions were filtered into round bottom flasks (rbf) and warmed once again gently to achieve dilute and clear solutions with no solid particles, thus to prevent self-seeding of the original form.6 The clear solutions in the rbf were dried rapidly using a rotovapor by setting an appropriate reduced pressure, water bath temperature (50 °C) and revolution speed of the rbf (130 rpm). Upon the completion of the solvent evaporation, a continuous vacuum was applied, in order to reach the minimum possible pressure, (9–10 mbar) and held there for about 5 min before collecting the dry solids for characterization. As the solvates can convert to other forms over time, the characterization was done immediately after their preparation without much delay. The phase purity of the solids was established by DSC and comparing the experimental PXRD patterns with those calculated from the corresponding single crystal data (Fig. S3†). IR spectroscopy was also performed using the same batches of samples (Fig. S1†).
Single crystal preparation
Succinylsulfathiazole drug was dissolved in the boiling solvents acetonitrile (MeCN), ethyl acetate (EA), water (H2O), acetone (AcMe) or tetrahydrofuran (THF). The resulting clear solutions were boiled for 10 min before being filtered into a fresh conical flask. The filtrate was left to evaporate slowly at ambient conditions. Single crystals suitable for X-ray diffraction studies were obtained in 4–6 days.
Powder X-ray diffraction (PXRD)
The PXRD patterns were collected on a Rigaku SmartLab with Cu Kα radiation (λ = 1.540 Å). The tube voltage and amperage were set at 40 kV and 50 mA, respectively. Each sample was scanned between 5 and 70° 2θ with a step size of 0.02°. The instrument was previously calibrated using a silicon standard.
Crystallography
Crystals of the SST forms were individually mounted on a glass pip. Intensity data were collected on a Bruker's KAPPA APEX II CCD Duo system with graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). All the data were collected at 100 K. Data reduction was performed using Bruker SAINT software.8a The crystal structures were solved by direct methods using SHELXL-97 and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for non-H atoms using SHELXL-97.8b Hydrogen atoms associated with carbon atoms were fixed in geometrically constrained positions. Hydrogen atoms associated with oxygen and nitrogen atoms were included in the located positions. Structure graphics shown in the figures were created using the X-Seed software package version 2.0.9
Differential scanning calorimetry (DSC)
DSC was conducted on a Mettler-Toledo DSI1 STARe instrument. Accurately weighed samples (2–3 mg) were placed in hermetically sealed aluminium crucibles (40 μL) and scanned from 30 to 300 °C at a heating rate of 5 °C min−1 under a dry nitrogen atmosphere (flow rate 80 mL min−1). The data were analyzed using STARe software.
Thermogravimetric analysis (TGA)
TGA was performed on a Mettler-Toledo TGA/SDTA 851e instrument. Approximately 10–15 mg of the sample was added to an aluminium crucible and heated from 30 to 500 °C at a rate of 10 °C min−1 under continuous nitrogen purging.
IR spectroscopy
Transmission infrared spectra of the solids were obtained using a Fourier-transform infrared spectrometer (PerkinElmer 502). KBr samples (2 mg in 20 mg of KBr) were prepared and 6 scans were collected at 4 cm−1 resolution for each sample. The spectra were measured over the range of 4000–400 cm−1.
Computational details
Geometry optimization of the SST tautomers was performed with Gaussian 0310 using the B3LYP method11 with the 6-31G(d) basis set,12 followed by single point energy calculations at the 6-311++G(2df, 2p) level, in a density functional theory (DFT) type calculation. The initial atomic coordinates for the molecules were always taken from the crystal structures.
2. Results and discussion
Screening by fast evaporation technique
Fast evaporation method was employed to conduct a screening for identifying possible new solid forms and to prepare all the known forms of SST from suitable solvents (Table S2†). In our recent reports we established the efficiency of the FE method for screening of co-crystals, co-crystal polymorphs7a and single component polymorphic systems.7b Here our reinvestigation of the old drug, succinylsulfathiazole, allowed us to test the utility of the FE method for identification and preparation of the API solvates (Table 1). Characterization of FE products by PXRD, IR spectroscopy, DSC and TGA confirmed the formation of pure solids of previously known polymorphic forms, SST-I and SST-II, a monohydrate and two new solvates, SST/AcMe and SST/THF.
Table 1 A comparison table of the present and previous crystallization studies employed for preparing various solid forms of succinylsulfathiazole
| Previous study |
Present study |
| Slow evaporation |
Fast evaporation |
| Used condition |
Resulted form |
Used condition |
Used condition |
Resulted form |
| Evaporation of AcMe solution to dryness on a water bath |
Polymorph I (earlier name, form I) |
Slow evaporation from MeCN at r.t. |
50 °C, 130 r.p.m., 250 mbar from EA solvent |
Polymorph I |
| — |
SST/AcMe (new) |
Slow evaporation from AcMe at r.t. |
50 °C, 130 r.p.m., 400 mbar from AcMe solvent |
SST/AcMe (new) |
| Suspension of any of the other crystal forms in water |
SST/2H2O (earlier name, form II) |
— |
50 °C, 130 r.p.m., 30 mbar from H2O solvent |
Mixture of SST/H2O and SST/2H2O |
| Crystallization from H2O, MeOH or EtOH (or) precipitation from a solution in NaOH by the addition of HCl |
SST/H2O (earlier name, form III) |
Crystallized from H2O, MeOH or EtOH |
50 °C, 130 r.p.m., 300 mbar from MeOH solvent |
SST/H2O
|
| Evaporate on a water bath of a AcMe solution until the first crystals separated, followed by evaporation at r.t. |
Polymorph II (earlier name, form IV) |
Slow evaporation from EA at r.t. |
50 °C, 130 r.p.m., 250 mbar from EtOH solvent |
Polymorph II |
| — |
SST/THF (new) |
Slow evaporation from THF at r.t. |
50 °C, 130 r.p.m., 250 mbar from THF solvent |
SST/THF (new) |
| Slow evaporation from n-butanol |
SST/But (earlier name form V) |
— |
— |
— |
| Slow evaporation from n-pentanol |
SST/Pnt (earlier name form VI) |
— |
— |
— |
Commercial SST sample contained the monohydrate form (SST/H2O). The FE product obtained from water had a mixture of two hydrated forms (Fig. S2a†) and also a possible unidentified form (in the PXRD two new peaks were observed at 27° and 29° which did not match with any known form). The PXRD pattern of SST/2H2O obtained by a slow evaporation method showed a good agreement with the previously identified dihydrate form (Fig. S2a†). As reported in previous studies SST formed cake like particles in water.13 Despite several attempts, we could not obtain single crystals suitable for SCXRD. It was also observed that all the forms of SST converted to SST/H2O on long exposure to the atmosphere (Fig. S4†), but not to SST/2H2O which is contrary to findings in previous reports. However, SST-II converted to SST/2H2O powder.
In the case of polymorphs, the solvent used was different for the slow and fast evaporation methods. The single crystals of SST-I and SST-II were obtained from MeCN and EA, respectively, by slow evaporation but for the same, EA and EtOH were used in the FE method. In case of the solvates, the same solvent was used for both slow and fast evaporation methods. The FE product from MeCN, a mixture of MeCN–MeOH, and MeOH resulted in only SST/H2O (Fig. S3b†). The two new solvate forms, SST/AcMe and SST/THF (see Fig. S1†), obtained by the FE method, could not be prepared by the liquid assisted grinding (LAG) method (Fig. S5†). LAG14 always resulted in SST/H2O. Probably, the intake of moisture during the LAG promoted the exclusive formation of SST/H2O. The results of the FE method suggest that a closed environment and faster kinetics during the evaporation of the solvent probably helped to prevent water intake, thus resulting in the solvates, instead of hydrates. This study proves the unique advantage of the FE method for quick screening of solvates. Hence the FE method, which is complementary to the existing screening techniques, has potential to become a regular screening tool in solid state pharmaceutical laboratories.
Crystal structure analysis
Single crystal structures were determined for the two polymorphic forms, SST-I and SST-II, and two solvates, SST/AcMe and SST/THF. The structure of the monohydrate (SST/H2O) is a redetermination. Crystallographic data are listed in Table 2.
Table 2 Crystallographic data and structure refinement parameters of different forms of SST
|
|
SST-I
|
SST-II
|
SST/H2O
|
SST/AcMe
|
SST/THF
|
| Chemical formula |
C13H13N3O5S2 |
C13H13N3O5S2 |
C13H13N3O5S2, H2O |
2(C13H13N3O5S2), C3H6O |
2(C13H13N3O5S2), C4H8O |
| Formula weight |
355.38 |
355.38 |
373.40 |
768.85 |
782.87 |
| Cryst sys |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P21/c |
P |
P |
|
a (Å) |
8.8478(4) |
11.351(3) |
16.190(6) |
8.5229(4) |
8.7032(2) |
|
b (Å) |
13.3523(6) |
8.577(2) |
5.1460(14) |
14.5887(7) |
14.5604(5) |
|
c (Å) |
14.1584(6) |
16.684(3) |
19.449(7) |
15.2327(7) |
15.5676(5) |
|
α (°) |
67.9870(10) |
90 |
90 |
64.7770(10) |
64.602(2) |
|
β (°) |
77.1140(10) |
107.483(5) |
94.028(17) |
78.1610(10) |
77.240(2) |
|
γ (°) |
74.6120(10) |
90 |
90 |
82.0860(10) |
83.635(2) |
| Vol (Å3) |
1480.77(11) |
1549.4(6) |
1616.4(9) |
1674.27(14) |
1737.79(9) |
|
D
calcd (g cm−3) |
1.594 |
1.523 |
1.534 |
1.525 |
1.496 |
|
μ (mm−1) |
0.390 |
0.373 |
0.365 |
0.353 |
0.342 |
|
θ range (°) |
1.57–27.00 |
1.88–27.00 |
1.26–26.50 |
1.50–25.14 |
1.48–26.96 |
|
Z
|
4 |
4 |
4 |
2 |
2 |
| Range h |
−11 to +11 |
−14 to +14 |
−20 to +19 |
−10 to +10 |
−11 to +11 |
| Range k |
−17 to +17 |
−10 to +10 |
−6 to +6 |
−17 to +17 |
−18 to +17 |
| Range l |
−18 to +13 |
−21 to +11 |
−24 to +24 |
−18 to +17 |
−19 to +17 |
| Reflns collected |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 736 736 |
14705 |
13143 |
24403 |
27707 |
| Independent reflns |
6458 |
3370 |
3322 |
5957 |
7527 |
| Obsd reflns |
5701 |
2241 |
2672 |
5375 |
5176 |
|
T (K) |
100 |
100 |
100 |
100 |
100 |
|
R
1
|
0.0344 |
0.0428 |
0.0391 |
0.0718 |
0.0440 |
|
wR
2
|
0.1132 |
0.1170 |
0.1016 |
0.1465 |
0.1250 |
| GOF |
0.873 |
0.895 |
1.018 |
1.044 |
0.882 |
| CCDC no. |
943847
|
943848
|
943849
|
943850
|
943851
|
A hydrogen bond table (Table S1†) and the ORTEP diagrams (Fig. S6–S10†) for all the solid forms are included in the ESI.† Examination of all the structures revealed a tautomeric preference in the polymorphs and pseudopolymorphs where the SST molecule adopts the imidine tautomeric form. The structure determination also allowed us to study the hydrogen bonding or synthon15 competition among various functional groups in the different forms.
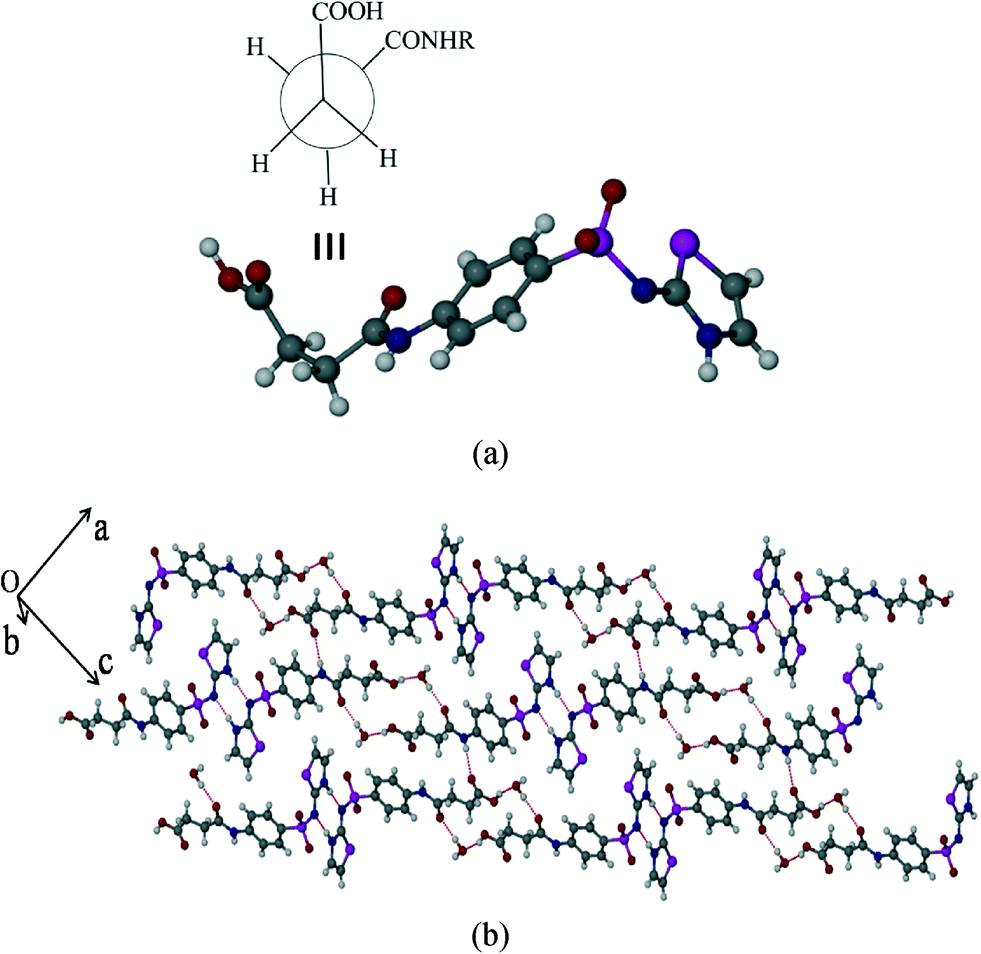
Polymorph-I (SST-I)
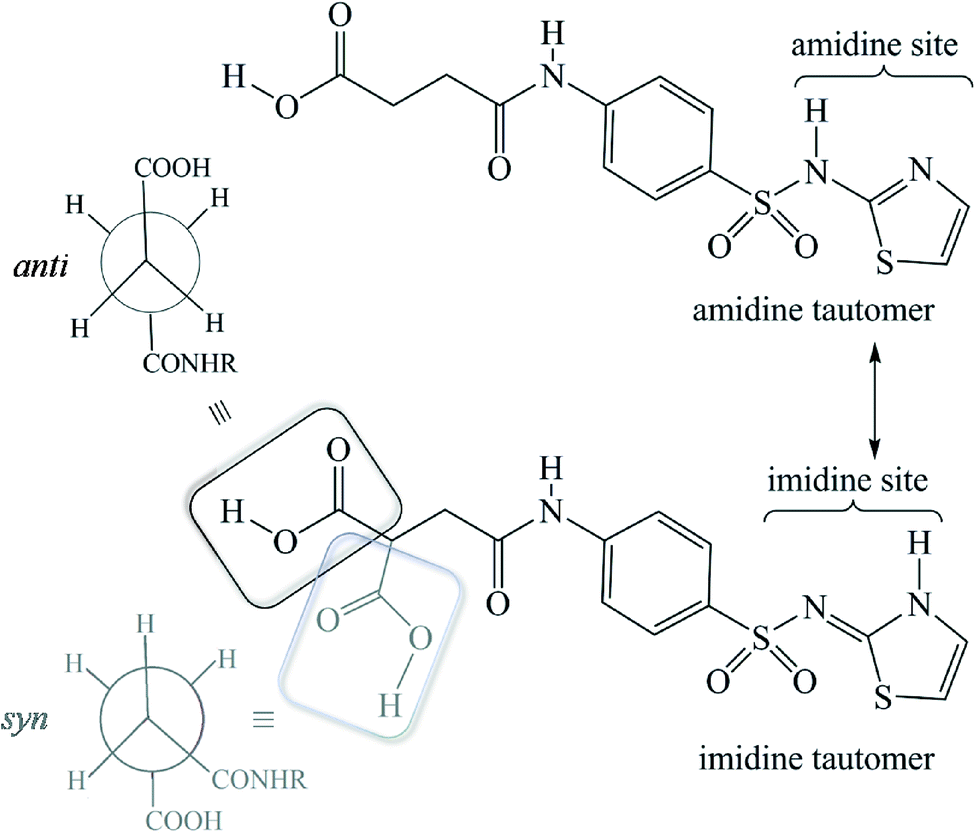
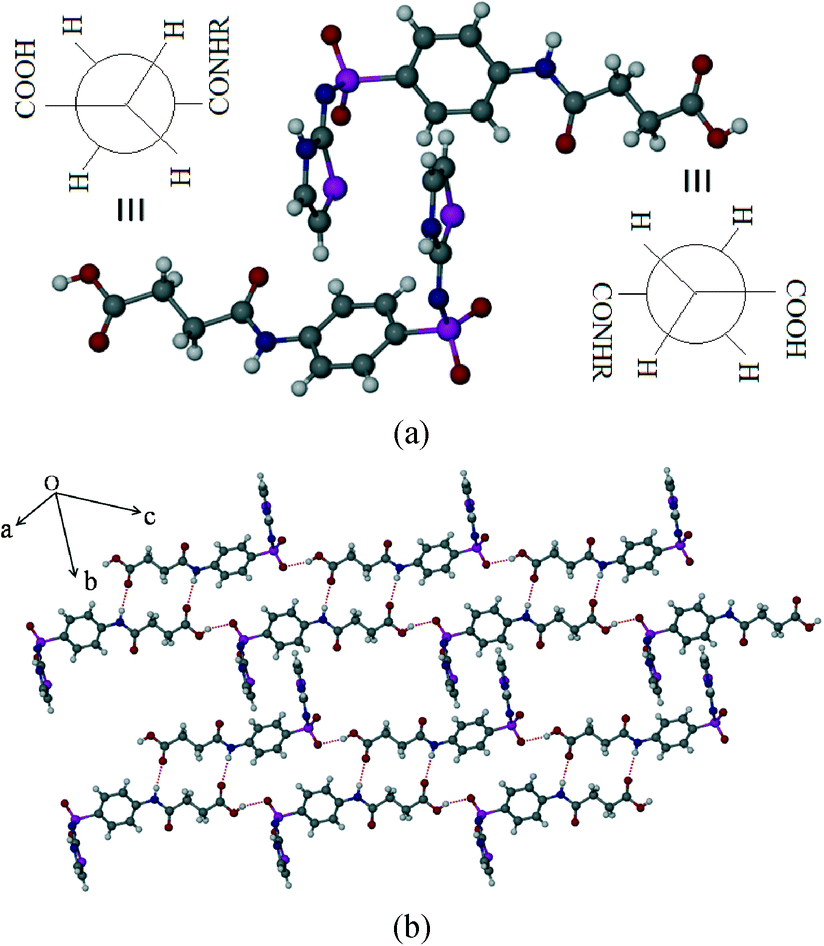
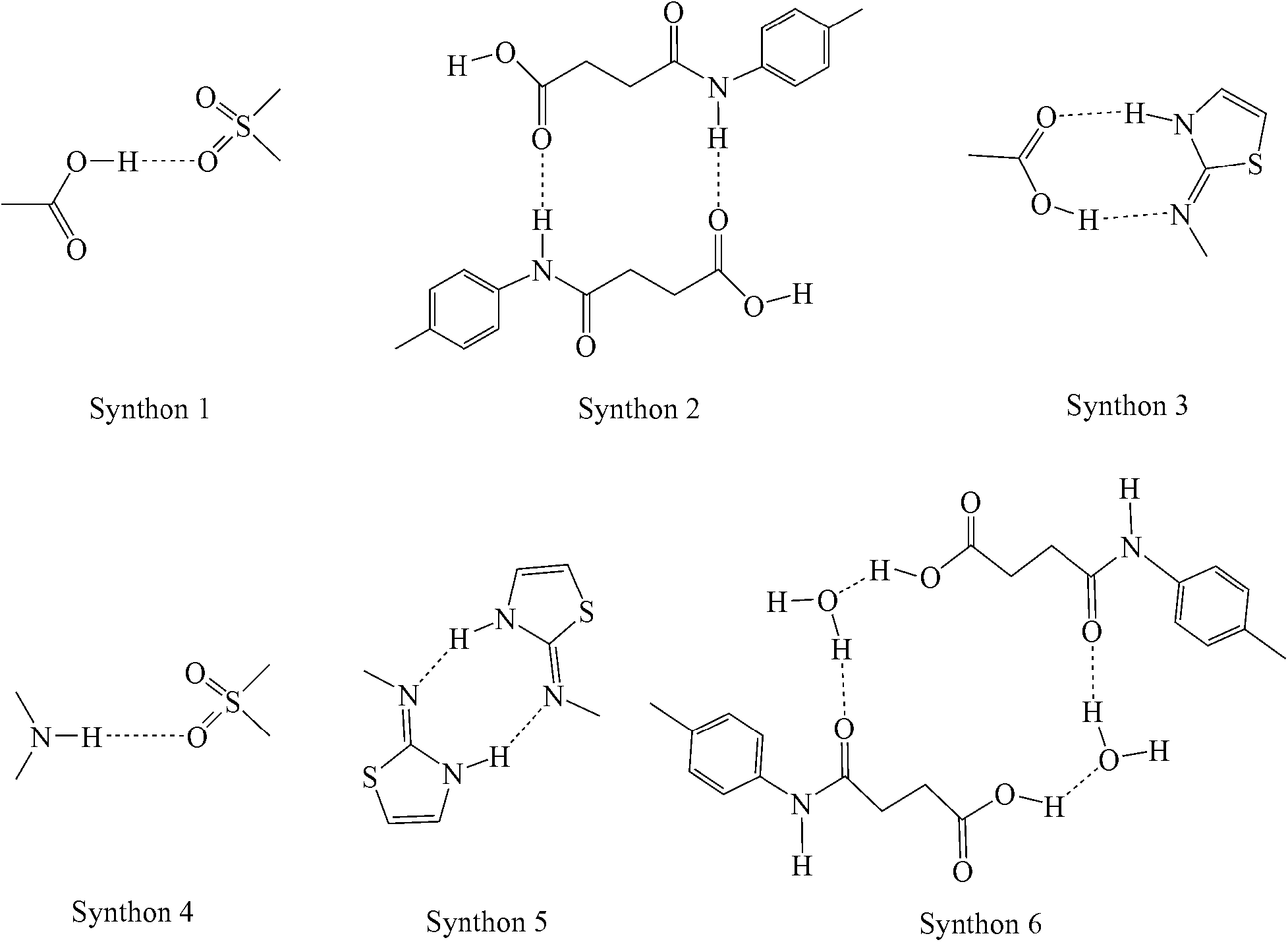
SST-I crystallizes in the triclinic P space group with two molecules in the asymmetric unit. Both the SST molecules adopt an L-shape conformation and exist in the imidine tautomeric form. Tautomerism, a common phenomenon in sulphonamide drugs,16–20 occurs via transfer of the proton from sulfonamide NH to the thiazole ring N as shown in Scheme 1. Notably, –CH2–CH2– of succinyl group in both the SST molecules adopts the anti geometry (Fig. 1). Two independent SST molecules interact via two separate O–H⋯O hydrogen bonds by involving the hydroxyl of CO2H and SO2 groups by synthon 1 (see Scheme 2; O(6)–H(6A⋯O(9); d/Å, θ/°: 1.9 Å, 160° and O(2)–H(2C)⋯O(4): 1.97 Å, 155°) to form linear chains which are further connected by (amide)N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(carboxyl) hydrogen bonds via synthon 2 (Scheme 2; N(4)–H(4)⋯O(1)C(1): 2.09 Å, 160° and N(1)–H(1)⋯O(7)C(14): 2.04 Å, 174°) leading to a ladder type network (Fig. 1b). Adjacent ladders arranged in an antiparallel fashion are connected via(imidine)N–H⋯OC(amide) hydrogen bonds. Because of the L-shape conformation of SST molecules, the thiazole rings lie nearly perpendicular to the direction of the ladder network. These thiazole rings from adjacent antiparallel ladders close pack as shown in Fig. 1a.
C(carboxyl) hydrogen bonds via synthon 2 (Scheme 2; N(4)–H(4)⋯O(1)C(1): 2.09 Å, 160° and N(1)–H(1)⋯O(7)C(14): 2.04 Å, 174°) leading to a ladder type network (Fig. 1b). Adjacent ladders arranged in an antiparallel fashion are connected via(imidine)N–H⋯OC(amide) hydrogen bonds. Because of the L-shape conformation of SST molecules, the thiazole rings lie nearly perpendicular to the direction of the ladder network. These thiazole rings from adjacent antiparallel ladders close pack as shown in Fig. 1a.
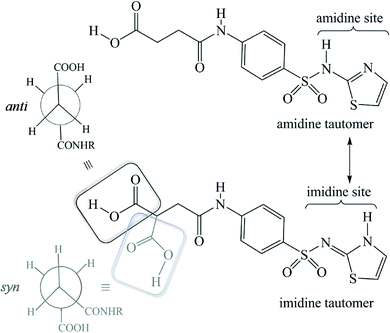 |
| | Scheme 1 Schematic representation of the tautomerism and conformational flexibility in succinylsulfamethazole (SST). Notice the alternate conformations, anti (black) or syn (grey) geometries, which can be adopted by the succinyl group. | |
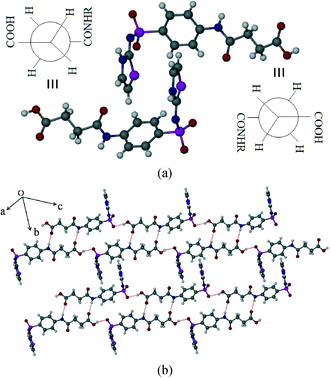 |
| | Fig. 1 Polymorph I of succinylsulfathiazole (SST-I). (a) L-shape molecular geometry of two independent SST molecules with the anti –CH2–CH2– conformation. (b) Ladders formed by the combination of synthons 1 and 2 (see Scheme 2). | |
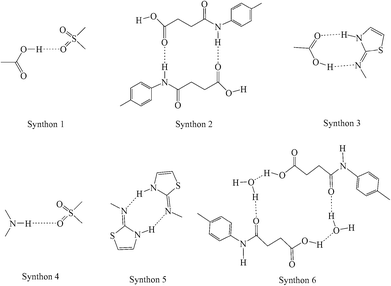 |
| | Scheme 2 Synthons observed in the SST polymorphs, hydrate and solvates. | |
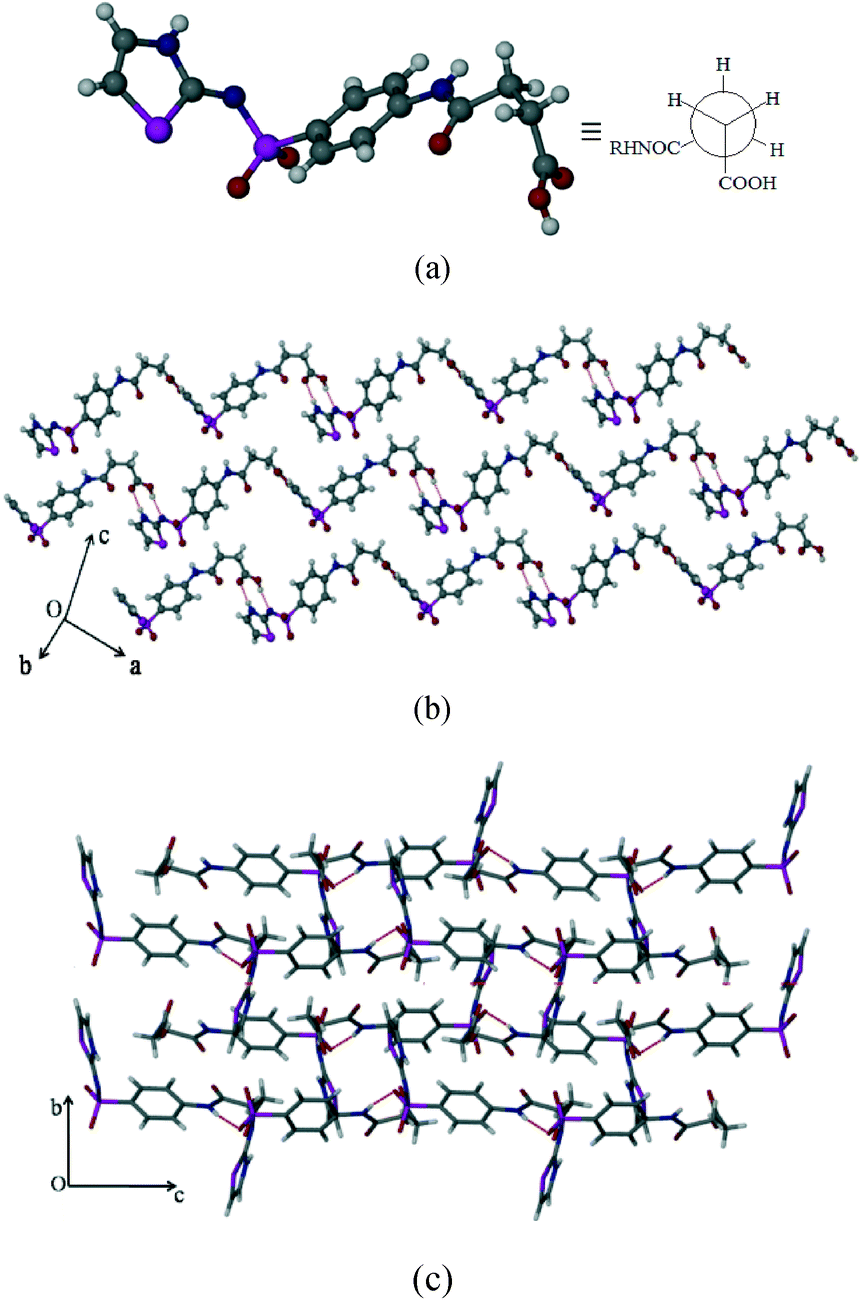
Polymorph-II (SST-II)
SST-II crystallizes in the monoclinic P21/c space group with one molecule in the asymmetric unit. The head-to-tail interaction by imidine and the carboxylic acid groups (N3–H3⋯O1, 1.96 Å, 174°; O2–H2C⋯N2, 1.94 Å, 166°) of the adjacent SST molecules via a heterodimer, synthon 3, forms wave like chains (Fig. 2b). Interestingly, the formation of the heterodimer is facilitated by the S-shape conformation of SST molecules due to the syn geometry of the –CH2–CH2– group. The parallel wave like chains are orthogonally connected by (sulfonamide)N–H⋯O(sulfoxy) hydrogen bonds via synthon 4 (N(1)–H(1)⋯O(5); 2.15 Å, 170°), leading to a 2D network. Thiazole rings of one 2D layer fill the voids in another adjacent layer resulting in interdigitation.
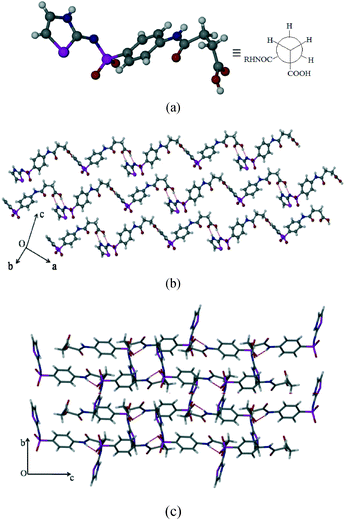 |
| | Fig. 2 Polymorph II of SST (SST-II). (a) Syn geometry of the SST at the –CH2–CH2– group. (b) Formation of wave like chains. (c) Interlocked 3D packing viewed along the a-axis. | |
Succinylsulfathiazole/monohydrate (SST/H2O)
The hydrated form of SST crystallizes in the monoclinic P21/c space group with one molecule of SST and a water molecule in the asymmetric unit. SST is in the syn or S-type conformation. Here the imidine sites of SST molecules primarily form a centrosymmetric homodimer by N–H⋯N hydrogen bonds via synthon 5 (N(3)–H(3)⋯N(2): 1.8 Å, 166°). The water molecules bridge the –CO2H and amide carbonyl O– atom via synthon 6 (O(1)–H(1A)⋯O(6): 1.74 Å, 158° and O(6)–H(6A)⋯O(3): 1.84 Å, 174°) as shown in Fig. 3b. The second proton of the water is linked to the sulfoxy group of the next layer, thus the strong hydrogen bonds are extended in all three dimensions.
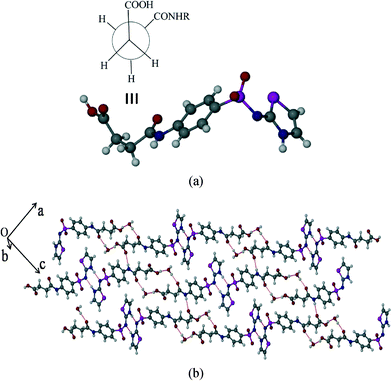 |
| | Fig. 3 SST/monohydrate (SST/H2O). (a) Syn geometry adopted at –CH2–CH2– group of SST. (b) Packing of two adjacent chains, showing the interactions formed by bridging water molecules between two SST molecules. | |
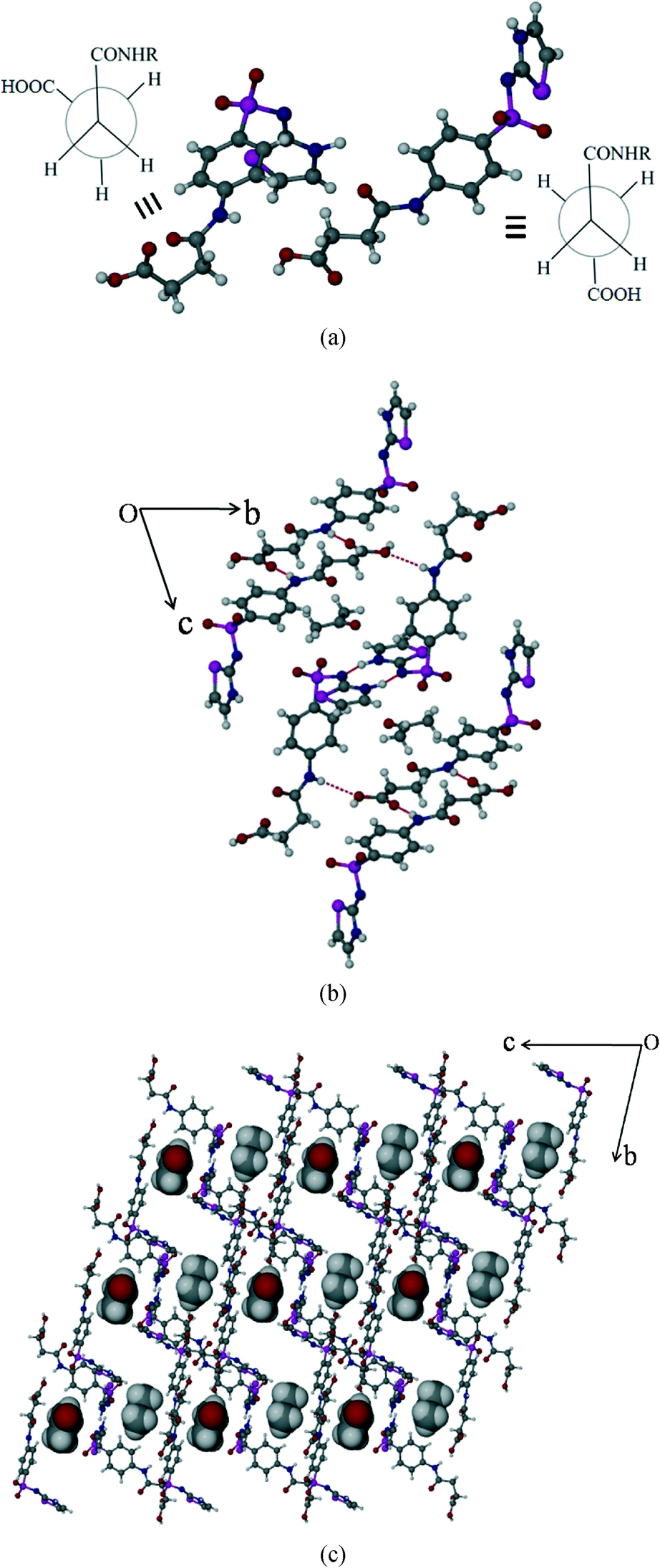
Succinylsulfathiazole/acetone solvate (SST/AcMe)
The SST/AcMe solvate crystallizes in the monoclinic P space group with two molecules of SST and one molecule of AcMe in the asymmetric unit. Interestingly, in this structure the two independent SST molecules adopt different conformations at the succinyl group; the first SST molecule is in the anti (L-shape) geometry whereas the second is in the syn (S-shape) geometry. Hence, both the conformational variants of SST are simultaneously seen in this structure. The SST molecules of the same symmetry form synthon 5 homodimers involving their imidine sites via N–H⋯N hydrogen bonds (N(3)–H(3)⋯N(2): 2.0 Å, 176° and N(6)–H(6)⋯N(5): 1.77 Å, 154°). The two different conformers facilitate the carboxylic acid groups to form different synthons. Two linear SST molecules are connected through (amide)N–H⋯OC(acid) hydrogen bonds, in an antiparallel fashion, via synthon 2 (N(1)–H(1A)⋯O(2)C(1): 2.1 Å, 171°). The leftover OH group of the acid is linked to the amide CO of the bent SST conformers. Whereas the acid OH of the bent SST molecule is linked to the amide CO of the linear SST to complete the loop by O–H⋯O [O(1)–H(1)⋯O(8): 1.9 Å, 159° and O(6)–H(6B)⋯O(3): 2.07 Å, 136°]. The solvent AcMe molecules are linked to the two S-shape SST dimers via bifurcated C–H⋯O interactions, and occupy channels formed by the host molecules along the a-axis (Fig. 4b).
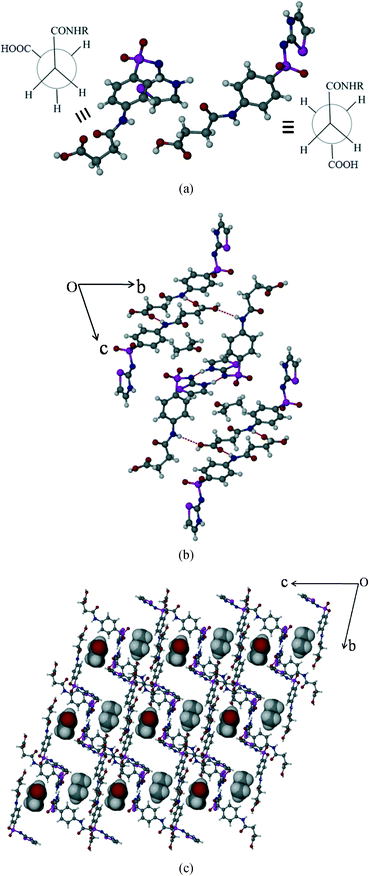 |
| | Fig. 4 Packing of SST/AcMe solvate. (a) Syn and anti geometries of two independent SST molecules at –CH2–CH2–. (b) Crystal packing showing the different types of interactions. (c) Occupation of solvent molecules, AcMe, in channels formed by host SST molecules. | |
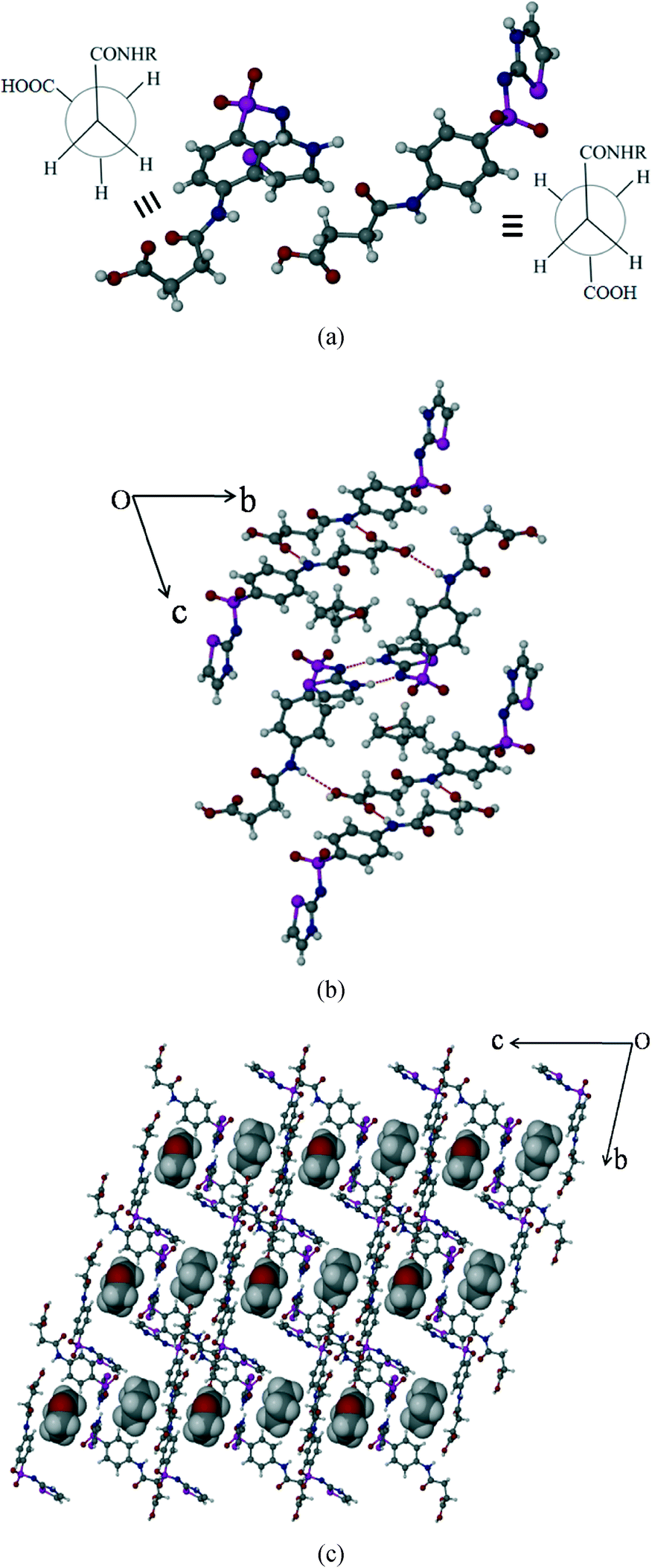
Succinylsulfathiazole/tetrahydrofuran solvate (SST/THF)
SST/THF also crystallizes in the triclinic P space group with two molecules of SST and one molecule of THF in the asymmetric unit. Indeed, this is isostructural16 to SST/AcMe, hence the synthons formed are the same, except some minor differences in the solvent THF interactions with the neighbouring molecules (Fig. 5b).
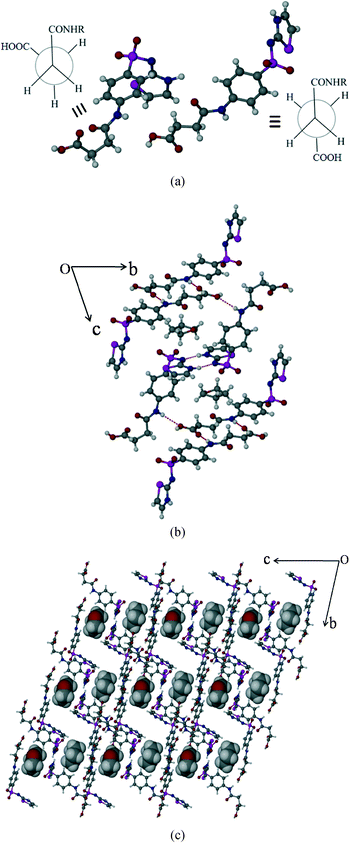 |
| | Fig. 5 Packing of SST/THF solvate. (a) Syn and anti geometries of two independent SST molecules at –CH2–CH2–. (b) Crystal packing showing the different types of interactions present in the structure. (c) Occupation of solvent molecules, THF, in channels formed by host SST molecules. | |
Tautomeric preference: density functional theory calculations
Both tautomerism and polymorphism are common phenomena in sulfonamide drugs.17–21 In this series of succinylsulfathiazole polymorphs and pseudopolymorphs, the API exists only in the imidine tautomeric form, in that the proton transfer occurs from the sulphonamide N-atom to the thiazole N-atom (Scheme 1). Hence, the amidine tautomeric form of SST is not seen in any of the structures, including in the earlier reported solvates of butanol and pentanol.6 A search in the Cambridge Structural Database (CSD) (V 5.34) for sulfonamide drugs, with at least >3 hits (exclusively) of the imidine tautomeric form, revealed that such a preference is present only in two other cases, sulfathiazole and sulfapyridine (Scheme S1†). To rationalize the tautomeric preference in the solid forms of SST, we performed DFT calculations using Gaussian software. In all cases, the molecules were taken from their corresponding crystal structures and a geometry optimization was performed, before proceeding to the single point energy calculations. The calculated energy difference between the amidine and corresponding imidine tautomeric forms revealed that the imidine tautomeric form is more stable than the amidine tautomeric form by 0.1816 kcal mol−1, which is consistent with the observed tautomeric preference of the former. It is to be noted that this energy difference between the two tautomeric forms corresponds to the isolated gaseous state molecules, but in the crystalline environment, with stabilizing hydrogen bonds, the difference could increase further to promote the imidine form. A more detailed computational study may unravel the fundamental reasons behind the imidine tautomeric preference in the solid forms of SST and other sulphonamide drugs.
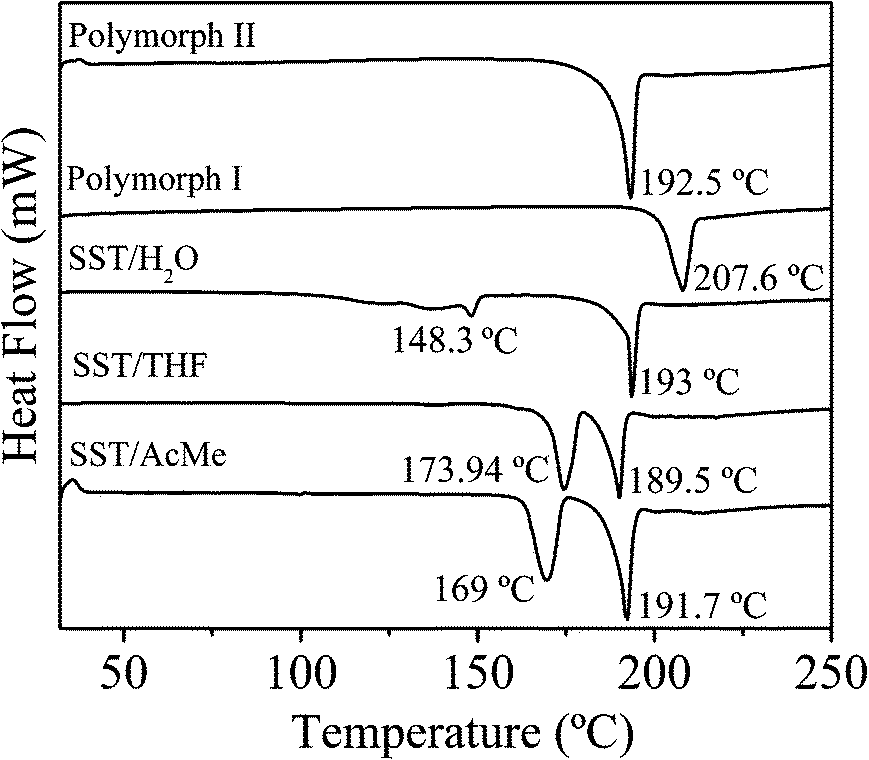
Thermal properties of the SST solid forms
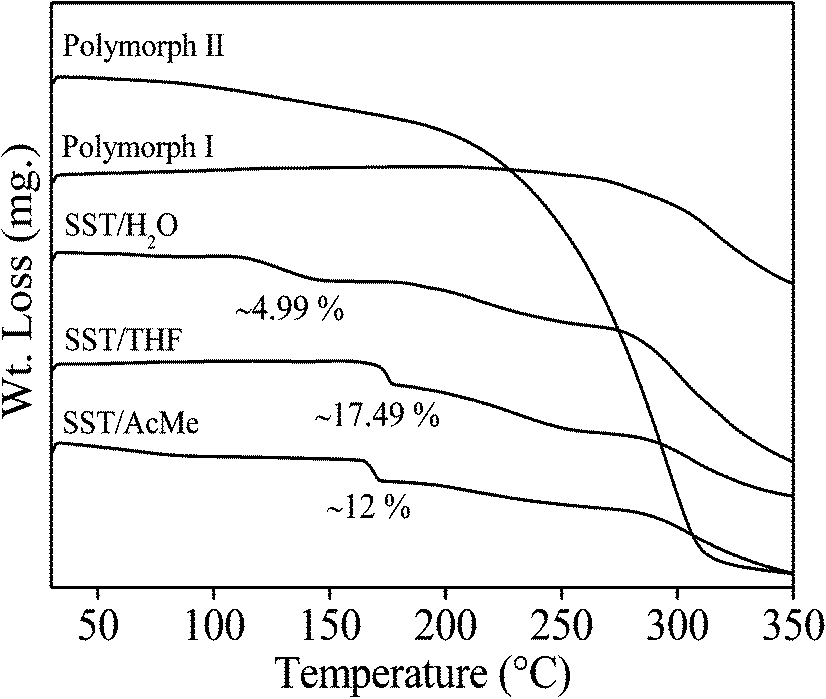
The thermal behaviour of all the polymorphs and pseudopolymorphs of SST, were studied by DSC and TGA experiments (Fig. 6 and 7). The DSC thermograms for SST-I and SST-II show a single endothermic transition peak each at 207.6 °C and 192.5 °C, respectively, corresponding to melting. The DSC thermogram of SST/H2O showed two endotherms; a minor peak at 148.33 °C, corresponding to the loss of a water molecule, and a major peak at 193.02 °C, corresponding to melting. The two solvates, SST/AcMe and SST/THF, also showed two endotherms each. Major endotherms corresponding to melting were observed at 191.7 °C and 189.5 °C for the SST/AcMe and SST/THF solvates, respectively, while minor endotherms appeared at 169.08 °C (for the loss of AcMe) and 173.94 °C (for the loss of THF). The analyses of the DSC results suggest that SST-I is thermodynamically more stable compared to all the other forms.
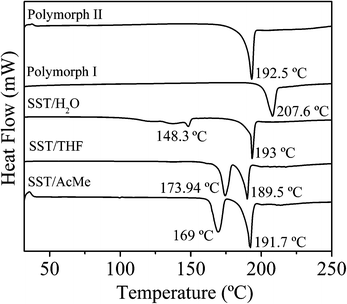 |
| | Fig. 6 Differential scanning calorimetry plots of different forms of SST. | |
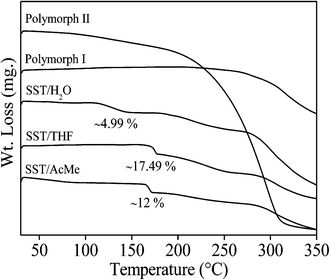 |
| | Fig. 7 Thermogravimetric analysis plots of different forms of SST. | |
The TGA experiments of the two polymorphic forms showed a single weight loss associated with decomposition after melting. In the case of hydrate and solvates, two weight losses were observed. For the SST monohydrate, the first small endotherm corresponding to loss of water (∼4.99% of the total weight) matched approximately to one mol of water. The slurry product of SST/H2O in water also showed a similar weight loss (4.82%), suggesting that there is no conversion of SST/H2O to SST/2H2O. The weight loss of the solid obtained from the slow evaporation method is (9.12%), equivalent to two moles of water (Fig. S2b†). For the SST/THF and SST/AcMe solvates, the first broad weight loss corresponding to the loss of solvent molecules (∼17.49% and ∼12.0% of the total weight, respectively) is equivalent to one mol of THF and AcMe, respectively.
Synthon competition study
SST has a total of three strong hydrogen bond donors, from its acid (–OH), amide (–NH) and imidine (–NH) groups, and five strong acceptors, namely from its acid (CO), amide (CO), SO2 (two SO) and imidine (N-atom) groups. Hence, there is an imbalance of acceptors and donors, which can potentially lead to multiple solid forms. Analysis of the synthons formed in the current and previously reported SST/But, SST/Pnt and SST/H2O suggests the following. The most popular interaction is synthon 5, a homodimer between the imidine groups, which is observed in all of the five solvates (or pseudopolymorphs), but absent in both the polymorphs. The second most popular is synthon 2, which is formed between the acid (CO) and amide (NH) groups when there is at least one SST conformer in the anti geometry. As can be expected, the most dominating groups are the imidine site and the carboxylic acid group. But synthon 3, formed between these two groups, is observed only once in SST-II. The remaining synthons 1, 4 and 6, which are seen occasionally, involve the less effective SO2 and amide (CO/NH) groups. However, notably, the most stable form, SST-I, with synthon 1 and 2, and anti geometry at the –CH2–CH2– groups, has the highest density of crystal packing. The balance among the large number of competing functional groups in a conformationally flexible molecule can easily be influenced by different solvent conditions. Hence the formation of several solid forms of SST is in line with general observations in the solid state pharmaceutical chemistry and crystal engineering.
Conclusions
Screening of succinylsulfathiazole by a fast evaporation (FE) method for possible new solid forms and to prepare all the known forms of SST from various solvents, demonstrated the efficiency of the technique for the identification and preparation of API solvates. Two new solvate forms, SST/AcMe and SST/THF were identified successfully by the FE method, which could not be obtained by the liquid assisted grinding (LAG) method. Probably, the intake of moisture during the LAG promotes the exclusive formation of SST/H2O. Hence the study demonstrates the complementary nature of the FE method to existing screening techniques and its potential to become a good screening tool in solid state pharmaceutical laboratories. In contrast to previous studies, we observed that the SST/H2O is more stable under ambient conditions than SST/2H2O. The DSC study revealed that SST-I is thermodynamically the more stable form compared to any other form. In this study, all the solid forms of SST were seen exclusively in the imidine form which was rationalized by performing DFT calculations. The energy difference between the two tautomeric forms is consistent with the imidine preference in the structures. Crystal structure analysis revealed that the homodimer formed by the imidine sites is the most popular synthon, which is observed in all of the SST solvates.
Acknowledgements
PPB thanks the CSIR for SRF. DST is gratefully thanked by CMR for financial support (SR/FT/CS-074/2009).
References
- G. L. Amidon, H. Lennernas, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413 CrossRef CAS.
- Process for Release of Biologically Active Species, P. Augustijns, J. A. Martens, R. Mellaerts and G. V. D. Mooter, Katholieke Universiteit Leuven, K. U. Leuven R & D, Patent application number: 20110060020, 03.10.2011. (http://www.faqs.org/patents/app/20110060020#b) Search PubMed.
- T. Hou, J. Wang, W. Zhang and X. Xu, J. Chem. Inf. Model., 2007, 47, 208 CrossRef CAS PubMed.
-
(a)
Method for Preparing Suspensions of Low-Solubility Materials, Publication number: US 2010/0247666 A1, Application number: 12/714577 Search PubMed;
(b)
Pharmaceutical compositions with improved dissolution, Publication number: WO2004000284 A1, Application number: PCT/US2003/019574 Search PubMed;
(c)
Polymer coated drug-ion exchange resins and methods, Publication number: US 2008/0118570 A1, Application number: 11/561,723 Search PubMed.
-
(a) M. A. Moustafa, S. A. Khalil, A. R. Ebian and M. M. Motaw, J. Pharm. Sci., 1974, 63, 1103 CrossRef CAS;
(b) A. R. Ebian, M. A. Moustafa, S. A. Khalil and M. M. Motaw, J. Pharm. Sci., 1975, 64, 1481 CrossRef CAS;
(c) M. A. Moustafa, A. R. Ebian, S. A. Khalil and M. M. Motaw, J. Pharm. Sci., 1975, 64, 1485 CrossRef.
- S. A. Bourne, M. R. Caira, L. R. Nassimbeni and I. Shabalala, J. Pharm. Sci., 1994, 83, 887 CrossRef.
-
(a) P. P. Bag, M. Patni and C. M. Reddy, CrystEngComm, 2011, 13, 5650 RSC;
(b) P. P. Bag and C. M. Reddy, Cryst. Growth Des., 2012, 12, 2740 CrossRef.
-
(a)
SAINT Plus (version 6.45), Bruker AXS Inc.: Madison, WI, 2003 Search PubMed;
(b)
SMART (version 5.625) and SHELX-TL (version 6.12), Bruker AXS Inc.: Madison, WI, 2000 Search PubMed.
-
L. J. Barbour, X-Seed, Graphical Interface to SHELX-97 and POV-Ray, University of Missouri, Columbia: Columbia, MO, 1999 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision E. 01, Wallingford CT: Gaussian, Inc., 2004 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef PubMed;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
-
(a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed;
(b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- N. Rodier, A. Chauvet and J. Masse, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 218 CrossRef.
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem. Commun., 2005, 880 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- Olanzapine drug shows isostructural monohydrate solvates of acetone, acetonitrile, nitromethane, and ethanol solvate form II. All the solvate structures crystallize in C2/c space group with similar unit cell parameters and are isostructural, R. Thakuria and A. Nangia, Cryst. Growth Des., 2013, 13, 3672 CAS.
-
(a) S. S. Yang and J. K. Guillory, J. Pharm. Sci., 1972, 61, 26 CrossRef CAS;
(b) J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS;
(c) M. C. Etter and J. C. Macdonald, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef;
(d) J. Grell, J. Bernstein and G. Tinhofer, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1030 CrossRef PubMed.
-
(a)
A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed;
(b) G. R. Desiraju and J. A. R. P. Sarma, Proc.–Indian Acad. Sci., Chem. Sci., 1986, 96, 599 CrossRef CAS.
- R. K. R. Jetti, R. Boese, J. A. R. P. Sarma, L. S. Reddy, P. Vishweshwar and G. R. Desiraju, Angew. Chem., Int. Ed., 2003, 42, 1963 CrossRef CAS PubMed.
-
(a) T. Gelbrich, T. L. Threlfall, A. L. Bingham and M. B. Hursthouse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o323 CAS;
(b) S. Ghosh, P. P. Bag and C. M. Reddy, Cryst. Growth Des., 2011, 11, 3489 CrossRef CAS;
(c) M. R. Caira, Mol. Pharmaceutics, 2007, 4(3), 310 CrossRef CAS PubMed.
-
(a) G. J. Kruger and G. Gafner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 326 CrossRef;
(b) G. J. Kruger and G. Gafner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 272 CrossRef CAS;
(c) M. M. Parmar, O. Khan, L. Seton and J. L. Ford, Cryst. Growth Des., 2007, 7, 1635 CrossRef CAS;
(d) T. Gelbrich, D. S. Hughes, M. B. Hursthouse and T. L. Threlfall, CrystEngComm, 2008, 10, 1328 RSC;
(e) P. McArdle, Y. Hu, A. Lyons and R. Dark, CrystEngComm, 2010, 12, 3119 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Geometrical parameters of molecules from crystal structures, powder X-ray diffraction patterns, infrared spectra, ORTEP diagrams. CCDC 943847–943851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce42159g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a