 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structures of benzoic acids with substituted pyridines and quinolines: salt versus co-crystal formation†
Gaëlle
Ramon
*,
Kate
Davies
and
Luigi R.
Nassimbeni
Supramolecular Chemistry Group, Department of Chemistry, University of Cape Town, Rondebosch, 7701, South Africa. E-mail: gaelle.ramon@uct.ac.za; Fax: +27 21 650 5768; Tel: +27 21 650 5184
First published on 13th January 2014
Abstract
A series of five substituted benzoic acids with 10 substituted pyridines and quinolines have been crystallized so that their ΔpKa, defined as pKa base–pKa acid, varied from −1.14 to +4.16. This spans the ‘uncertainty’ region for the formation of salt versus co-crystals. Although most of our results confirmed that structure formation of co-crystal versus salt parts at ΔpKa ≈ 2, we report here a structure that does not follow the general rule and serves as a cautionary tale.
Introduction
The question of pH control in the formation of salts versus co-crystals is a topic of current research interest. This is particularly important in the pharmaceutical industry which is continually seeking Active Pharmaceutical Ingredients (APIs) with desirable properties such as stability and taste.When an acid is reacted with a base, the ensuing product will be a salt or a co-crystal, and the general rule is that if ΔpKa (pKa base–pKa acid) is greater than 2–3, the product will be a salt.1 This is not an absolute rule, because the pKa is an acid dissociation constant measured in water at a fixed temperature. However, the product of the reaction depends upon the solvent or mixture of solvents, the temperature and, if the product is a crystalline solid, on the packing forces impinging on the molecules or ions in the crystal structure.
However the ΔpKa can be used as a guideline and may be justified by a simplified analysis of the equilibria between a carboxylic acid RCOOH and a substituted pyridine base R′Py.
If the equilibrium constants of the base and the acid are Ka1 and Ka2 respectively:
| RCOOH ↔ RCOO− + H+ |

| R′PyH+ ↔ R′Py + H+ |
Here we have approximated concentration with activities. For the reaction:

| RCOOH + R′Py → RCOO− + R′PyH+ |
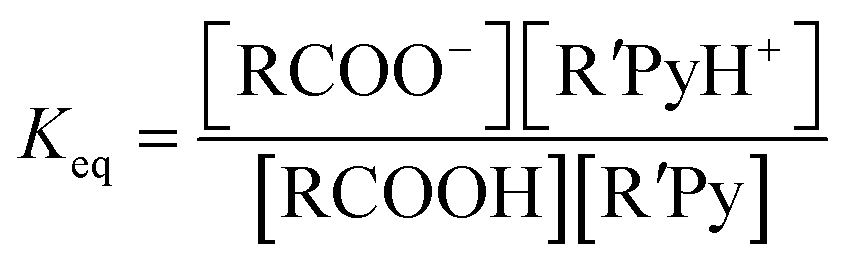
and

If ΔpKa = 0 there are equal quantities of ions and neutral molecules in the solution.
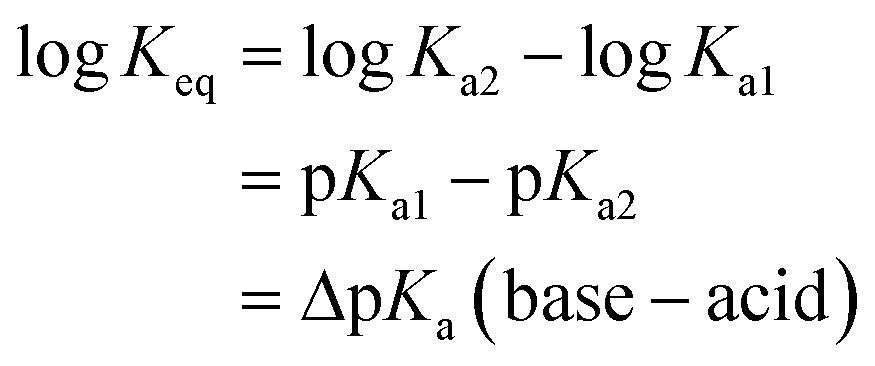
If ΔpKa > 0, the ionic species will predominate and vice versa.
For example, if pKa1 = 5 and pKa2 = 3, ΔpKa = +2 so Keq ≈ Ka2/Ka1 = 10−3/10−5 ≈ 100. Thus the concentration of the ionized species is in excess, giving a greater likelihood of the salt being formed.
In the crystalline solid state, the difference between a salt and a co-crystal is subtle, being dependent in the unambiguous location of a hydrogen atom as shown in Fig. 1.
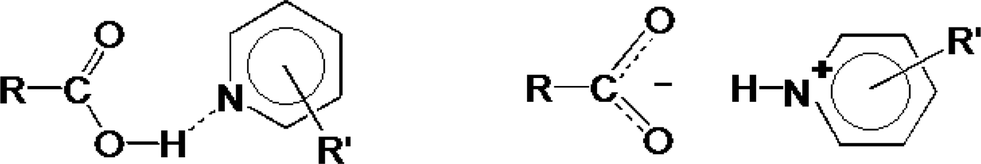 | ||
| Fig. 1 Co-crystal versus salt formation. | ||
Thus if the O⋯N distance is typically ≈2.7 Å and O–H and N+–H distances are ≈1.0 Å, the movement of the H atom is small, approximately 0.7 Å.
Fig. 1, however displays schematically the extreme, either–or situation. It has been pointed out by Childs, Stahly and Park2 that there are cases where the location of the proton is ambiguous and is not clearly covalently bonded to either the oxygen or the nitrogen atom. The proton transfer should thus be regarded as a continuum and can, in addition, be traced by the difference in bond lengths in the carboxyl group.
These are distinctly different in the acid (d C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ≈ 1.2 Å, d C–OH ≈ 1.30 Å) but tend to equalize in the carboxylate anion (d C–O ≈ 1.26 Å).
O ≈ 1.2 Å, d C–OH ≈ 1.30 Å) but tend to equalize in the carboxylate anion (d C–O ≈ 1.26 Å).
Recently, Cruz-Cabeza3 carried out a survey of over 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures comprising ionised and non-ionised acid-base pairs. She found a linear relationship between ΔpKa and the probability of proton transfer, in the pKa range of −1 to +4. The cross-over point occurs at ΔpKa = 1.3, beyond which there is a greater than 50% probability of the product being a salt. Gilli & Gilli4 have devised a slide rule which predicts the strength of a Donor–Acceptor hydrogen bond based on the ΔpKa of the system. It is noteworthy that their device predicts salts if ΔpKa > 3, co-crystals when ΔpKa < −3 and the uncertainty region which contains the strong hydrogen bonds is pictured as D⋯H⋯A, which may be regarded as the true three-centre-four-electron system.
000 structures comprising ionised and non-ionised acid-base pairs. She found a linear relationship between ΔpKa and the probability of proton transfer, in the pKa range of −1 to +4. The cross-over point occurs at ΔpKa = 1.3, beyond which there is a greater than 50% probability of the product being a salt. Gilli & Gilli4 have devised a slide rule which predicts the strength of a Donor–Acceptor hydrogen bond based on the ΔpKa of the system. It is noteworthy that their device predicts salts if ΔpKa > 3, co-crystals when ΔpKa < −3 and the uncertainty region which contains the strong hydrogen bonds is pictured as D⋯H⋯A, which may be regarded as the true three-centre-four-electron system.
The Gilli & Gilli review points out that the interval of ΔpKa matching should be shifted by 1.5 units when interpreting crystal structures,5 which agrees with the findings in the survey by Cruz-Cabeza.
Based on the above, we have crystallized a series of five substituted benzoic acids with 10 substituted pyridines and quinolines as shown in Fig. 2.
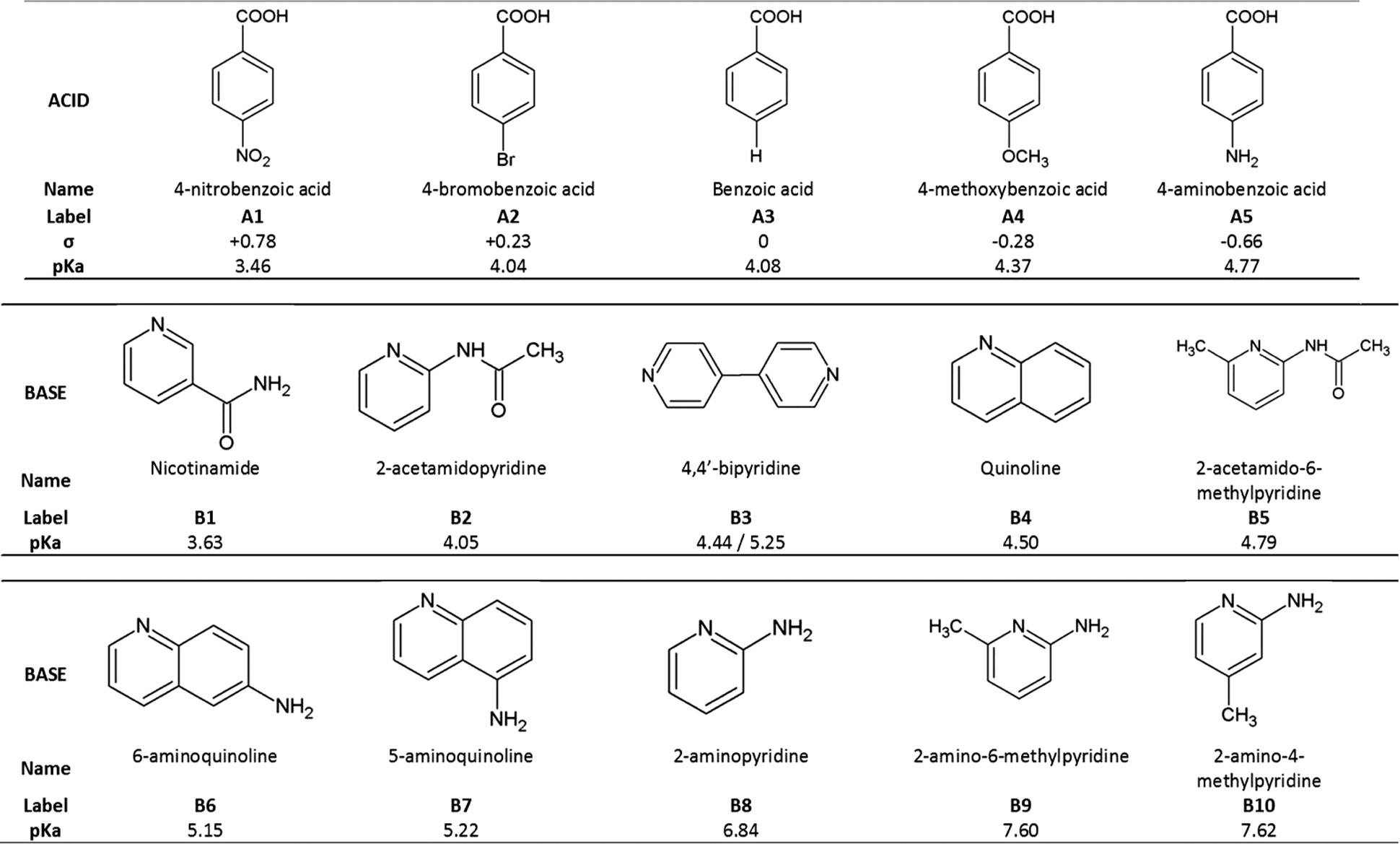 | ||
| Fig. 2 List of acids and bases used in this project and their pKa. | ||
The acids were selected to have different Hammett substituent constants varying from p-amino (σ = −0.66) to p-nitro (σ = +0.78), yielding different pKa values,6 while the pKa values of the substituted bases varied from 3.63 to 7.62.
The ΔpKa values of our chosen compounds varied from −1.14 to +4.16, thus spanning the ‘uncertainty’ region identified by both Cruz-Cabeza and Gilli & Gilli.
In this work we discuss the bonding of 22 structures made up of acid-base pairs which either form salts or co-crystals. They give rise to 26 ΔpKa due to some bases having two pKa values. In Table 3 we label these 1 to 26. Eight structures8–23 were taken from the CSD7 while the remainder were newly elucidated and are labelled I to XIV as shown in Fig. 3.
 | ||
| Fig. 3 22 structures (14 new) illustrate the formation of co-crystals versus salts as a function of the ΔpKa (base–acid). In the case of bases with two pKa, the ΔpKa was calculated for both, hence the repeats in the table, for example: A4·B3 has two ΔpKa at 0.07 and 0.88. | ||
Table 1 for the co-crystals and Table 2 & 3 for the salts report the crystal data, structure and refinement parameters for I to XIV.
| Co-crystal | A5·B2 | A3·B2 | A5·B5 | 2A4·B3 | A1·B4 |
|---|---|---|---|---|---|
| I(1) | II(4) | III(5) | IV(6) | VI(12) | |
| ΔpKa = −0.72 | ΔpKa = −0.03 | ΔpKa = 0.02 | ΔpKa = 0.07 | ΔpKa = 2.46 | |
| Molecular formula | C7H7NO2· | C7H6O2· | C7H7NO2· | 2(C8H8O3)· | C7H5NO4· |
| C7H8N2O | C7H8N2O | C8H10N2O | C10H8N2 | C9H7N | |
| Molecular mass | 273.29 | 258.27 | 287.32 | 460.47 | 296.28 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | Pna21 | P21/n | P21/c | P21/c | P21 |
| a, Å | 14.864(1) | 12.776(3) | 8.435(2) | 9.0319(1) | 3.759(1) |
| b, Å | 18.554(2) | 5.184(1) | 23.547(5) | 10.860(1) | 27.082(9) |
| c, Å | 4.709(1) | 19.547(4) | 7.127(1) | 23.561(2) | 6.551(2) |
| α, deg | 90 | 90 | 90 | 90 | 90 |
| β, deg | 90 | 103.08(3) | 90.22(3) | 97.950(2) | 90.735(8) |
| γ, deg | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 1298.8(2) | 1261.1(5) | 1415.5(5) | 2288.8(3) | 666.9(4) |
| Z | 4 | 4 | 4 | 4 | 2 |
| D c, Mg m−3 | 1.398 | 1.360 | 1.348 | 1.336 | 1.475 |
| μ, mm−1 | 0.101 | 0.097 | 0.096 | 0.096 | 0.108 |
| F(000) | 576 | 544 | 608 | 968 | 308 |
| θ min–θmax, deg | 1.8–25.2 | 4.1–27.8 | 3.5–27.9 | 1.8–28.3 | 1.5–26.5 |
| Index ranges min./max. h,k,l | −17:17 |
−16:16 |
−11:11 |
−7:12 |
−4:4 |
| −22:22 |
−6:6 |
−30:23 |
−14:14 |
−33:34 |
|
| −5:5 |
−25:25 |
−9:6 |
−31:31 |
−8:8 |
|
| Reflections collected | 12991 |
4851 | 7340 | 31250 |
5055 |
| Independent reflections | 2330 | 2852 | 3315 | 5695 | 2702 |
| R int | 0.047 | 0.027 | 0.075 | 0.040 | 0.039 |
| Data/parameters refined | 2330/198 | 2852/182 | 3315/208 | 5695/317 | 2702/204 |
| No. of reflections with I > 2σ(I) | 2032 | 1873 | 1563 | 4225 | 1894 |
| Goodness of fit, S | 1.04 | 0.97 | 0.99 | 1.03 | 0.99 |
| R (F) [I > 2σ(I)] | 0.0329 | 0.0401 | 0.0583 | 0.0398 | 0.0470 |
| Final wR2 (all data) | 0.0788 | 0.1076 | 0.1566 | 0.1133 | 0.1029 |
| Largest diff. peak and hole, e Å−3 | −0.19, 0.12 | −0.23, 0.25 | −0.29, 0.25 | −0.22, 0.25 | −0.21, 0.16 |
| Salts | A5·B6 | A3·B8 | A2·B8 | A5·B10 | A4·B9 |
|---|---|---|---|---|---|
| V(8) | VII(16) | VIII(17) | IX(18) | X(19) | |
| ΔpKa = 0.38 | ΔpKa = 2.76 | ΔpKa = 2.80 | ΔpKa = 2.85 | ΔpKa = 3.23 | |
| Molecular formula | C7H6NO2· | C7H5O2· | C7H4BrO2· | C7H6NO2· | C8H7O3· |
| C9H9N2 | C5H7N2 | C5H7N2 | C6H9N2 | C6H9N2 | |
| Molecular mass | 281.31 | 216.24 | 295.13 | 245.28 | 260.29 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | Pbca | P21/c | P212121 | P21/n |
| a, Å | 7.335(2) | 11.500(2) | 9.686(1) | 5.554(1) | 9.440(2) |
| b, Å | 9.187(2) | 15.191(3) | 10.333(1) | 8.553(2) | 11.509(2) |
| c, Å | 21.307(4) | 12.228(2) | 12.002(1) | 25.380(5) | 12.690(3) |
| α, deg | 90 | 90 | 90 | 90 | 90 |
| β, deg | 93.30(3) | 90 | 97.765(2) | 90 | 110.77(3) |
| γ, deg | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 1433.3(5) | 2136.2(7) | 1190.2(1) | 1205.7(4) | 1289.2(5) |
| Z | 4 | 8 | 4 | 4 | 4 |
| D c, Mg m−3 | 1.304 | 1.345 | 1.647 | 1.351 | 1.341 |
| μ, mm−1 | 0.089 | 0.094 | 3.444 | 0.094 | 0.095 |
| F(000) | 592 | 912 | 592 | 520 | 552 |
| θ min–θmax, deg | 2.4–27.5 | 3.8–27.9 | 2.1–26.4 | 2.9–25.3 | 2.9–25.4 |
| Index ranges min./max. h,k,l | −9:9 |
−14:14 |
−12:12 |
−6:6 |
−11:11 |
| −11:11 |
−19:19 |
−12:12 |
−10:10 |
−13:13 |
|
| −27:27 |
−14:15 |
−15:15 |
−30:30 |
−15:15 |
|
| Reflections collected | 6289 | 4451 | 18549 |
2199 | 4578 |
| Independent reflections | 3266 | 2433 | 2439 | 2199 | 2355 |
| R int | 0.014 | 0.050 | 0.032 | 0 | 0.036 |
| Data/parameters refined | 3266/210 | 2433/157 | 2439/198 | 2199/184 | 2355/186 |
| No. of reflections with I > 2σ(I) | 2867 | 1432 | 2182 | 1468 | 1445 |
| Goodness of fit, S | 1.06 | 0.95 | 1.04 | 0.94 | 0.96 |
| R (F) [I > 2σ(I)] | 0.0363 | 0.0443 | 0.0208 | 0.0389 | 0.0423 |
| Final wR2 (all data) | 0.1032 | 0.1106 | 0.0540 | 0.0796 | 0.0972 |
| Largest diff. peak and hole, e Å−3 | −0.19, 0.28 | −0.25, 0.22 | −0.39, 0.26 | −0.22, 0.16 | −0.24, 0.19 |
| Salts | A4·B10 | A3·B9 | A2·B9·2(H2O) | A2·B10 |
|---|---|---|---|---|
| XI(20) | XII(22) | XIII(23) | XIV(24) | |
| ΔpKa = 3.25 | ΔpKa = 3.52 | ΔpKa = 3.56 | ΔpKa = 3.58 | |
| Molecular formula | C8H7O3· | C7H5O2· | C7H4BrO2· | C7H4BrO2· |
| C6H9N2 | C6H9N2 | C6H9N2·2(H2O) | C6H9N2 | |
| Molecular mass | 260.29 | 230.26 | 345.15 | 309.15 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | P21/c | Pca21 | P21/n | P21/c |
| a, Å | 10.337(2) | 11.888(2) | 17.124(2) | 9.579(2) |
| b, Å | 11.727(2) | 12.313(3) | 4.132(1) | 11.539(2) |
| c, Å | 11.031(2) | 16.815(3) | 22.882(2) | 11.311(2) |
| α, deg | 90 | 90 | 90 | 90 |
| β, deg | 105.53(3) | 90 | 111.762(2) | 100.45(3) |
| γ, deg | 90 | 90 | 90 | 90 |
| V, Å3 | 1288.4(4) | 2461.3(9) | 1503.5(2) | 1229.4(4) |
| Z | 4 | 8 | 4 | 4 |
| D c, Mg m−3 | 1.342 | 1.243 | 1.525 | 1.670 |
| μ, mm−1 | 0.096 | 0.085 | 2.748 | 3.339 |
| F(000) | 552 | 976 | 704 | 624 |
| θ min–θmax, deg | 3.5–27.9 | 2.9–28.0 | 1.9–26.4 | 4.1–27.9 |
| Index ranges min./max. h,k,l | −13:12 |
−13:13 |
−19:21 |
−10:12 |
| −11:14 |
−15:15 |
−5:5 |
−11:14 |
|
| −13:13 |
−21:21 |
−28:19 |
−14:12 |
|
| Reflections collected | 5041 | 4641 | 8753 | 6862 |
| Independent reflections | 2887 | 4641 | 3036 | 2789 |
| R int | 0.036 | 0 | 0.028 | 0.027 |
| Data/parameters refined | 2887/186 | 4641/333 | 3036/204 | 2789/176 |
| No. of reflections with I > 2σ(I) | 1640 | 3081 | 2400 | 2326 |
| Goodness of fit, S | 0.94 | 1.02 | 1.00 | 1.05 |
| R (F) [I > 2σ(I)] | 0.0462 | 0.0525 | 0.0303 | 0.0242 |
| Final wR2 (all data) | 0.1205 | 0.1297 | 0.0821 | 0.0556 |
| Largest diff. peak and hole, e Å−3 | −0.29, 0.26 | −0.24, 0.21 | −0.40, 0.59 | −0.53, 0.49 |
Experimental section
Crystal growth
The acid and base to be combined were selected and equal molar amounts were weighed accurately. The compounds were dissolved in separate vials in ethanol with gentle stirring and heating. The solutions were filtered and combined. The acid-base mixtures were allowed to crystallise at room temperature upon slow evaporation of the solvent to afford crystals suitable for single crystal structure determination.Crystal structure
For structures II, III, VII, X, XI, XII and XIV, the intensity data measured on a Bruker KAPPA CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) were used to determine the cell dimensions. The strategy for the data collections was evaluated using COLLECT software.23 For these structures, the intensity data were collected by the standard φ scan and omega scan techniques, scaled and reduced using the program DENZO-SMN.24Crystal structure determinations for I, IV, V, VI, VIII, IX and XIII were performed by single crystal X-ray diffraction using a Bruker KAPPA APEX II DUO diffractometer with graphite monochoromated Mo-Kα radiation. Unit cell refinement and data reduction were performed using the program SAINT.25
All structures were determined at low temperature (173 K) and the refinement parameters are presented in Tables 1–3.
Structures VII and IX were previously reported in the literature15–17 but for data collections at room temperature (VII and IX) and 208 K (for VII).
The structures were solved using direct methods and refined by full-matrix least squares with SHELX-97,26 refining on F2. The program X-Seed27 was used as a graphical interface. For all the structures the non-hydrogen atoms were found in the difference electron density map. The aromatic and methyl hydrogen atoms were placed in calculated positions and refined isotropically using a riding model. The hydrogen atoms belonging to amine, protonated pyridyl or hydroxyl groups were placed using the electron density map and refined isotropically. At times, the N–H distance in amines was restricted to an acceptable value using DFIX. We were careful to establish whether the compound was a co-crystal or a salt. In the case of co-crystals, the hydroxyl hydrogen atom was located in a difference density map and refined freely. In the case of salts, we located the hydrogen atom as being bonded to the N+ of the base and in addition we noted the equalisation of the C–O bond length of the carboxyl moiety.
The structures were deposited at the Cambridge Crystallographic Data Centre and allocated the numbers: CCDC 963007–963012, 963014–963021.
Results and discussion
Crystal structures of the co-crystals
Structure I, derived from 4-aminobenzoic acid and 2-acetaminopyridine, A5·B2, crystallises in the space group Pna21 with Z = 4. Each 4-aminobenzoic acid is stabilised by three hydrogen bonds as shown in Fig. 4, comprising a closed ring and an infinite chain running along [100]. In all figures, the same nomenclature has been employed whereby the acid molecules are always represented in orange while the base molecules display the conventional colours. Employing the graph nomenclature of Etter and Bernstein28 these may be designated as R22(8) and C22(12) respectively. In this packing the chains are anti-parallel running along [100] and are hydrogen bonded via a weak (Acid)–N–H⋯N–(Acid) displayed in blue in Fig. 4. The metrics of the hydrogen bonding for this and the remaining structures are reported in Table 4. | ||
| Fig. 4 Packing of structure I displaying anti-parallel ribbons running along [100]. The ribbons are weakly connected via (Acid)–N–H⋯N–(Acid) displayed in blue. | ||
|
Structure II consists of benzoic acid and 2-acetaminopyridine, A3·B2, in the space group P21/n with Z = 4. The structure is made of acid-base pairs which are hydrogen bonded with R22(8) motif similar to that of the previous structure.
Structure III of 4-aminobenzoic acid and 2-acetamido-6-methylpyridine, A5·B5, crystallises in space group P21/c with Z = 4. The structure is similar to that of I with the only difference arising from the 6-methyl group substitution on the base. The packing is still characterised by a chain of hydrogen bonds, C22(12) , running along [010], as well as the ring formed by the interaction of the carboxylic moiety with the amino-pyridine nitrogens of the base.
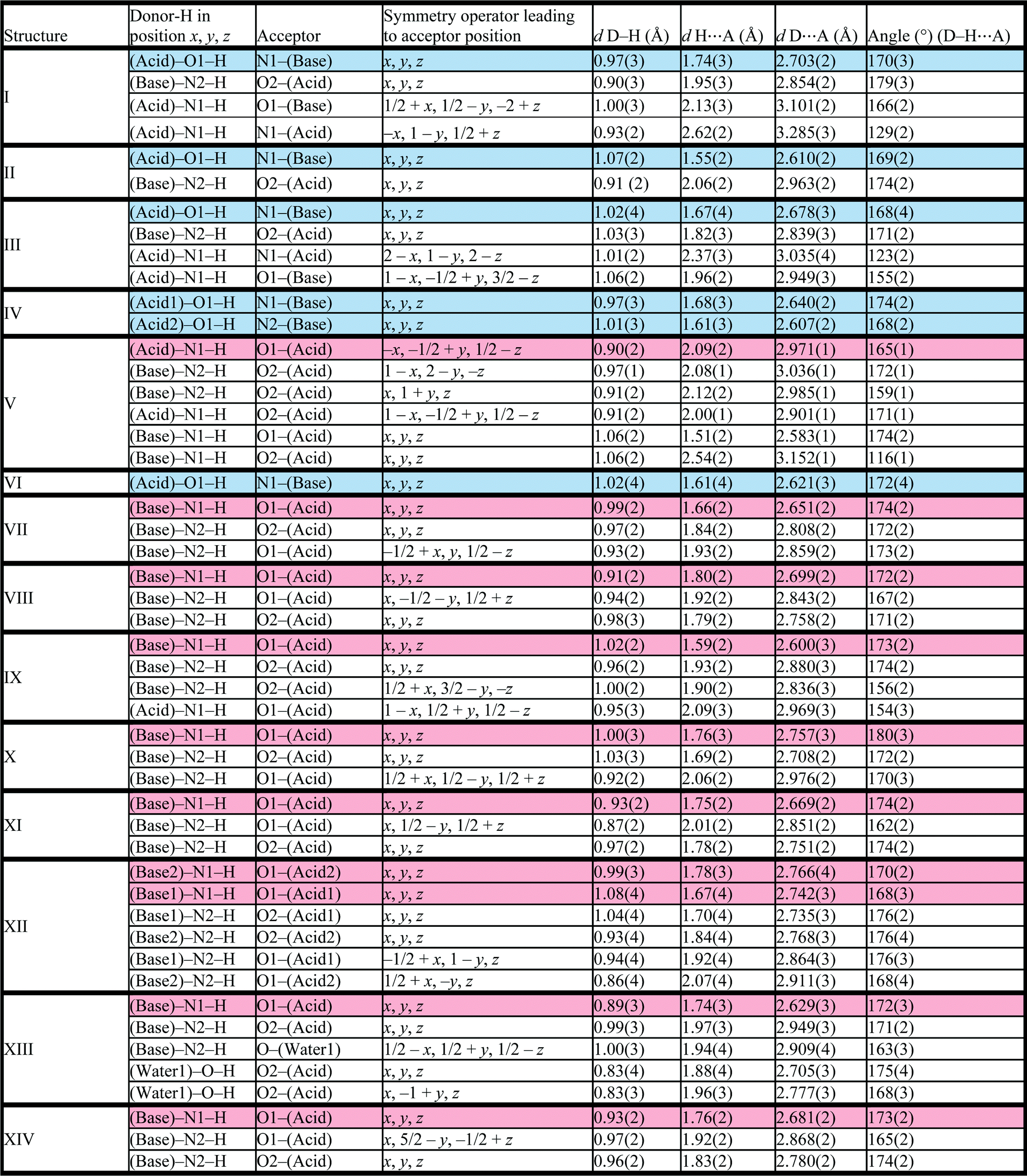
Structure IV arose from a mixture of 4-methoxybenzoic acid and 4,4′-bipyridine, 2A4·B3 and crystallises in P21/c with Z = 4. The bipyridine is di-basic and therefore has two distinct pKa values, giving rise to two points on the co-crystal/salt versus ΔpKa diagram reported in Fig. 3. The asymmetric unit consists of the bipyridine moiety hydrogen bonded to two 4-methoxybenzoic acids which may be represented by D22(10) bonds (Fig. 5).
 | ||
| Fig. 5 Hydrogen bonding motif in structure IV. | ||
The packing is characterised by columns of 4-methoxybenzoic acids and bipyridine running in the [108] direction.
Structure VI comprising 4-nitrobenzoic acid and quinoline, A1·B4, crystallises in space group P21 with Z = 2, the asymmetric unit displays the acid hydrogen bonded to the quinoline base via (Acid)O–H⋯N(Base). The packing is characterised by layers approximately parallel to the bc face, and stacked perpendicular to the [104] direction.
Crystal structures of the salts
Structure V from 4-aminobenzoic acid and 6-aminoquinoline, A5·B6, crystallises in space group P21/c with Z = 4. The asymmetric unit comprises one aminoquinolinium hydrogen bonded to an aminobenzoate. An inspection of Fig. 3 makes structure V appear as an outlier. However, the ΔpKa value of +0.38 yields a probability of salt formation of 34% as proposed by the Cruz-Cabeza assessment. In addition, this system is further stabilised by a complex hydrogen bonding network. In Fig. 6a, we show a central aminobenzoate anion which is hydrogen bonded to four neighbouring aminobenzoates and three aminoquinolinium cations. We note that there are five unique additional hydrogen bonds stabilising the structure as listed in Table 4. The packing shown in Fig. 6b displays alternate chains of anti-parallel aminobenzoates interleaved with aminoquinolates. | ||
| Fig. 6 Motif in structure V: a) a central aminobenzoate ion (in orange) is hydrogen bonded to seven ions-four aminobenzoates and three aminoquinoliniums, b) anti-parallel chains of aminobenzoate ions (in orange and pink) in the packing of V. | ||
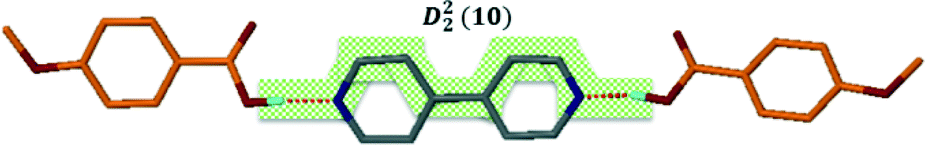
Structure VII is derived from benzoic acid and 2-aminopyridine, A3·B8. It crystallises in Pbca with Z = 8 and its packing is characterised by infinite chains which may be described as C22(6) R22(8) which run parallel to [100] (Fig. 7).
 | ||
| Fig. 7 Hydrogen bond motifs in structure VII displaying chains which run along [100]. | ||
Structure VIII arose from 4-bromobenzoic acid and 2-aminopyridine, A2·B8 and crystallises in the space group P21/c with Z = 4. The hydrogen bonding is comparable to that of structure VII with the infinite chains running parallel to [001].
Structure IX derives from 4-aminobenzoic acid and 2-amino-4-methylpyridine, A5·B10. The packing is characterised by a chain of hydrogen bonds, C33(14) R22(8) running along [001] which cross a spiral of hydrogen bonds C24(8) which extend along [110] (Fig. 8).
 | ||
| Fig. 8 Structure IX displaying the different hydrogen bond motifs with two types of chains running a) along [001] and b) along [110]. | ||
Structure X is derived from 4-methoxybenzoic acid and 2-amino-6-methylpyridine, A4·B9. It crystallises in P21/n and the packing displays C22(6) R22(8) similar to that of structure VII.
Structure XI crystallises from 4-methoxybenzoic acid and 2-amino-4-methylpyridine, A4·B10, in space group P21/c with Z = 4 and displays the same hydrogen bond motif as structure VII.
Structure XII was derived from benzoic acid and 2-amino-6-methylpyridine, A3·B9. It crystallises in the space group Pca21 with Z = 8. The packing is characterised by two crystallographically independent chains of hydrogen bonded ions. These C22(6) R22(8) chains run parallel to [100].
Structure XIII arose from 4-bromobenzoic acid and 2-amino-6-methylpyridine, A2·B9·2(H2O). This compound crystallises as a di-hydrate in the space group P21/n with Z = 4. One of the waters of crystallisation acts as a hydrogen bonding bridge creating a spiral of O–H⋯O bonds about the 2-fold screw axis at Wyckoff position e. In addition, the amino groups are hydrogen bonded to the oxygen of this water molecule. We again have the synthon of hydrogen bonds between the carboxylate moiety and the aminopyridinium cation. We may therefore describe this as two parallel chains running along [010] linked by ring systems. The motif resembles a ladder: C12(4) R46(12). The second water molecule links ladders together, giving rise to hydrogen bonded sheets which run along [010] (Fig. 9).
 | ||
| Fig. 9 Packing of the only solvated structure (XIII) obtained during this study: a) Packing of XIII viewed down [010] displaying sheets of hydrogen bonded molecules and solvents which run along [101]. b) Schematic representation of the ladders where only the moieties involved in hydrogen bonding are displayed. The ladders are connected through ribbons of hydrogen bonded water molecules running along [010]. | ||
Structure XIV arose from 4-bromobenzoic acid and 2-amino-4-methylpyridine, A2·B10, crystallising in space group P21/c with Z = 4. The packing motif is again C22(6) R22(8) similar to that of structure VII.
Conclusions
Fig. 3 shows that the break from co-crystal to salt occurs at ΔpKa ≈ 2. Structure 8(V), obtained from combining 4-aminobenzoic acid and 6-aminoquinoline, forms a salt. Its value of ΔpKa is +0.38, and is clearly an outlier (Fig. 3). However, according to the Cruz-Cabeza survey, there is a 34% chance of the compound being a salt at this value of ΔpKa, and the survey points out that secondary hydrogen bonds can influence the stabilisation of the ionic structure.29 This is the case with structure 8(V) in which, in addition to the (quinolinium)–N+–H⋯O−–CO–(benzoate) hydrogen bond, the amino–benzoate anion is further stabilised by six other N–H⋯O–CO hydrogen bonds, the metrics of which are given in Table 4.The hydrogen-bonding table is arranged so that each of the structures I to XIV displays the main H-bond (Acid)–O1–H⋯N1–(Base) in blue for the co-crystals or as (Protonated Base)–N1–H⋯O1–(Anion) in red for a salt. One notes that most of the structures listed in Table 4 display additional hydrogen bonds, a feature which is obviously common on the systems under study.
Acknowledgements
The authors would like to thank the South African National Research Foundation (NRF Pretoria) for the financial support.Notes and references
- P. H. Stahl and C. G. Wermuth, in Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-vch, 2002 Search PubMed.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmacol., 2007, 4, 323 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC.
- (a) P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed; (b) P. Gilli and G. Gilli, in Supramolecular Chemistry, from Molecules to Nanomaterials, ed P. A. Gale and J. W. Steed, Wiley, Chichester, 2012, vol. 6, ch. 2 Search PubMed.
- P. Huyskens, L. Sobczyk and I. Majerz, J. Mol. Struct., 2002, 615, 61–72 CrossRef CAS.
- J. Shorter, in Correlation Analysis in Organic Chemistry, Clarendon Press, Oxford, 1973 Search PubMed.
- Cambridge Structural Database, version 5.34 update Feb 2013 Search PubMed.
- B. Lou and S. Hu, J. Chem. Crystallogr., 2011, 41, 1663–1668 CrossRef CAS.
- D. E. Lynch, S. Chatwin and S. Parsons, Cryst. Eng., 1999, 2(2–3), 137–144 CrossRef CAS.
- R. Wang, F. Jiang, Y. Zhou, L. Han and M. Hong, Inorg. Chim. Acta, 2005, 358, 545–554 CrossRef CAS PubMed.
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2009, 9(2), 1106–1123 CAS.
- J. R. Bowers, G. W. Hopkins, G. P. A. Yap and K. A. Wheeler, Cryst. Growth Des., 2005, 5(2), 727–736 CAS.
- T.-F. Tan, J. Han, M.-L. Pang, H.-B. Song, Y.-X. Ma and J.-B. Meng, Cryst. Growth Des., 2006, 6(5), 1186–1193 CAS.
- J. A. Bis and M. J. Zaworotko, Cryst. Growth Des., 2005, 5(3), 1169–1179 CAS.
- M. Odabasoglu, O. Buyukgungor and P. Lonnecke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o51–o52 Search PubMed.
- D.-H. He, Y.-Y. Di, W.-Y. Dan, Y.-P. Liu and D.-Q. Wang, Acta Chim. Slov., 2010, 57, 458 CAS.
- H. Shen, J.-J. Nie and D.-J. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1129 CAS.
- S. R. Jebas and T. Balasubramanian, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o2209–o2211 CAS.
- T.-F. Tan, J. Han, M.-L. Pang, H.-B. Song, Y.-X. Ma and J.-B. Meng, Cryst. Growth Des., 2006, 6(5), 1186–1193 CAS.
- D. E. Lynch, Private Communication, 2009 Search PubMed.
- W.-M. Dai, H. Zhou and Y.-Q. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o578 CAS.
- M. Hemamalini and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o335 CAS.
- COLLECT, Data Collection Software, Nonius, Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Macromolecular Crystallography, ed. C. W. Carter, Jr and R. M. Sweet, Academic Press, 1997, part A, vol. 276, pp. 307–326 Search PubMed.
- SAINT, Version 7.60a, Bruker AXS Inc, Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, SHELX-97: Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, X-Seed: a Software Tool for Supramolecular Crystallography, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- (a) M. C. Etter and J. C. MacDonald, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef; (b) M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS; (c) M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS; (d) J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- V. Balevicius, R. Barisevicinte, K. Aidas, I. Svoboda, H. Ehrenberg and H. Fuess, Phys. Chem. Chem. Phys., 2007, 3181–3189 RSC.
Footnote |
| † The structures were deposited at the Cambridge Crystallographic Data Centre and allocated the numbers: CCDC 963007–963012, 963014–963021. CIF files have been submitted as ESI. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41963k |
| This journal is © The Royal Society of Chemistry 2014 |