
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directionality of π-holes in nitro compounds†
Antonio
Bauzá
a,
Tiddo J.
Mooibroek
*b and
Antonio
Frontera
 *a
*a
aDepartment of Chemistry, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma (Baleares), Spain. E-mail: toni.frontera@uib.es; Fax: +34 971 173426
bSchool of Chemistry of the University of Bristol, Cantock's Close, BS8 1TS, Bristol, UK. E-mail: chtjm@bristol.ac.uk
First published on 1st December 2014
Abstract
A statistical survey of the Cambridge Structural Database reveals that the interaction between the π-holes of nitro groups and electron-rich atoms is somewhat directional. High-level ab initio computations indicate energies up to −6.6 kcal mol−1.
Intermolecular interactions determine how molecules interact with one another, and are thus fundamental to inquiries in areas like supramolecular chemistry and molecular biology.1 It is well-appreciated that even weak interactions (e.g., hydrogen bonding involving C–H) can bear functional relevance, especially when several such forces work in concert.2 This begs the question what weak interactions can be identified as possibly functionally relevant.
Hydrogen (H) and halogen (Hlg) bonds are by far the best established intermolecular forces.3 The regions of electropositive potential on R–H and R–Hlg (Fig. 1a and b, phenol and bromobenzene, respectively) can be described as unpopulated σ* antibonding orbitals along the R–H/Hlg vector.7b,c These ‘σ-holes’ have also been identified on atoms that belong to the oxygen, nitrogen and carbon families, and their binding to electron-rich entities is referred to as chalcogen-,4 pnicogen-,5 and tetrel-bonding,6 respectively.
 | ||
| Fig. 1 Some σ-and π-holes with their electropositive potential in kcal mol−1 (MP2/6-311+G**). | ||
In analogy to the σ-hole, a π-hole can be seen as an electropositive potential located on an unpopulated π*-orbital. The best-known π-hole interactions involve carbonyl compounds (e.g., p-quinone, as shown in Fig. 1c): Bürgi and Dunitz7 uncovered the trajectory along which a nucleophile attacks the π-hole of a carbonyls' C-atom, and π-hole interactions involving amides are known to persist in protein structures.8 Aromatic rings bearing electron withdrawing substituents, such as hexafluorobenzene (Fig. 1d), can also function as π-holes.9 Interactions involving such π acidic aromatic rings are generally referred to as anion- or lone pair-π interactions.10
A recent theoretical study revealed that a π-hole can also be found on a nitro group.11 Association of this π-hole with electron-rich entities (ElR, e.g., H2O and Cl−) was estimated to vary in strength between −0.5 and −27.9 kcal mol−1. Fig. 1e and f illustrate the π-holes in nitromethane and nitrobenzene, respectively. One might thus wonder if the ‘NO2π⋯ElR’ interaction bears any functional relevance. The first step towards answering this question is to establish whether this intermolecular force is actually directional. Analysis of the data compiled within the Cambridge Structural Database (CSD)12 has proven to be an exquisite tool for revealing the directional nature of traditional and non-canonical intermolecular forces. We thus set out to evaluate the CSD using a methodology that is particularly apt for ascertaining the directional character of weak intermolecular forces.9
Aliphatic and aromatic C–NO2 compounds that are abundant within the CSD were considered: there are 900 crystallographic information files (CIFs) containing nitromethane, and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 227 CIFs containing an aromatic nitro compound where the NO2 moiety is flanked by two ortho-H's (this allows the NO2 and C6H2 moieties to be relatively coplanar). Fig. 2a illustrates the query used to obtain initial datasets from the CSD.
227 CIFs containing an aromatic nitro compound where the NO2 moiety is flanked by two ortho-H's (this allows the NO2 and C6H2 moieties to be relatively coplanar). Fig. 2a illustrates the query used to obtain initial datasets from the CSD.
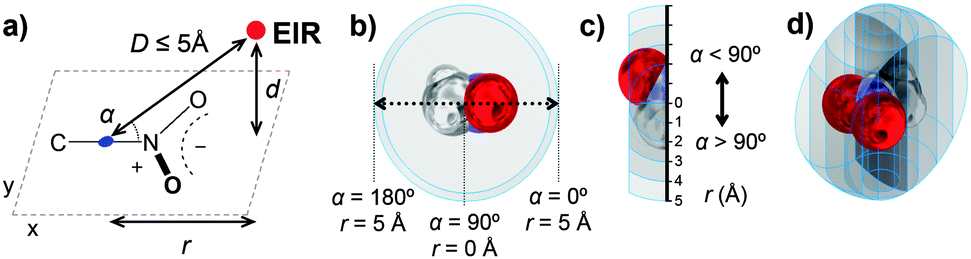 | ||
| Fig. 2 Illustrations of the method used to analyse directionality of intermolecular interactions between an electron-rich atom (ElR., N, P, As, O, S, Se, Te, F, Cl, Br, I or At) and a nitro group (in this illustration nitromethane). | ||
An entry was considered a hit when the intermolecular distance (D) between the C–N centroid (blue) and any electron-rich atom (red, ElR = N, P, As, O, S, Se, Te, F, Cl, Br, I or At) was ≤5 Å. The initial dataset was thus confined within a sphere with a radius of 5 Å, centred on the C–N centroid. The spatial separation (d) between ElR and the CNO2 plane was also retrieved from the CSD. The xy coordinates relative to this plane were determined as described elsewhere.13 By virtue of Pythagoras's theorem, the parallel displacement parameter r could be derived from D and d. The CNcentroid–N–ElR angle (α) was measured as well.
For this study, only the data characterized by y = ±2 Å were considered, which are therefore located within the 4 Å wide spherical segment, as illustrated in Fig. 2b–d. The data can thus be characterized by the parallel displacement (r) within this spherical segment, either towards the NO2 group (α < 90°) or towards the other atoms attached to C (α > 90°). To analyse directionality, we applied a method described in detail elsewhere.9,13,14 This method entails computing the parameter P(r),‡ which signifies the distribution of the data along r that is corrected for the volume occupied by the host and for a random scattering of data. When P ≠ 1, non-accidental clustering of data is established and P > 1 is indicative of an attractive interaction.
The P versus r plots for CH3NO2 (black circles) and C6H2NO2 (open circles) are shown on the left hand side of Fig. 3. For both ‘hosts’, P > 1 around r = 0, indicating some directionality of the π-hole. The P-values of 2–3 are similar to those observed for C–H⋯πphenyl hydrogen bonding.15 In the case of CH3NO2, the observed clustering is spread out over the region ranging from r = −1 to +1 Å. It is likely that CH⋯ElR hydrogen bonding also contributes to the observed distribution. The clustering observed for aromatic NO2 is spread out over the region from r = 0 to +1 Å, and hydrogen bonding cannot contribute in this case. It was further assessed how exactly the data are distributed within the data characterized by −1 > r < 1 Å. Thus, the relative hit fraction was plotted as a function of the van der Waals corrected CNcentroid⋯ElR distance, as shown on the right hand side of Fig. 3. For both central groups, roughly 20% van der Waals overlap is present. These two plots together indeed show that a nitro moiety can be a directional π-hole donor.
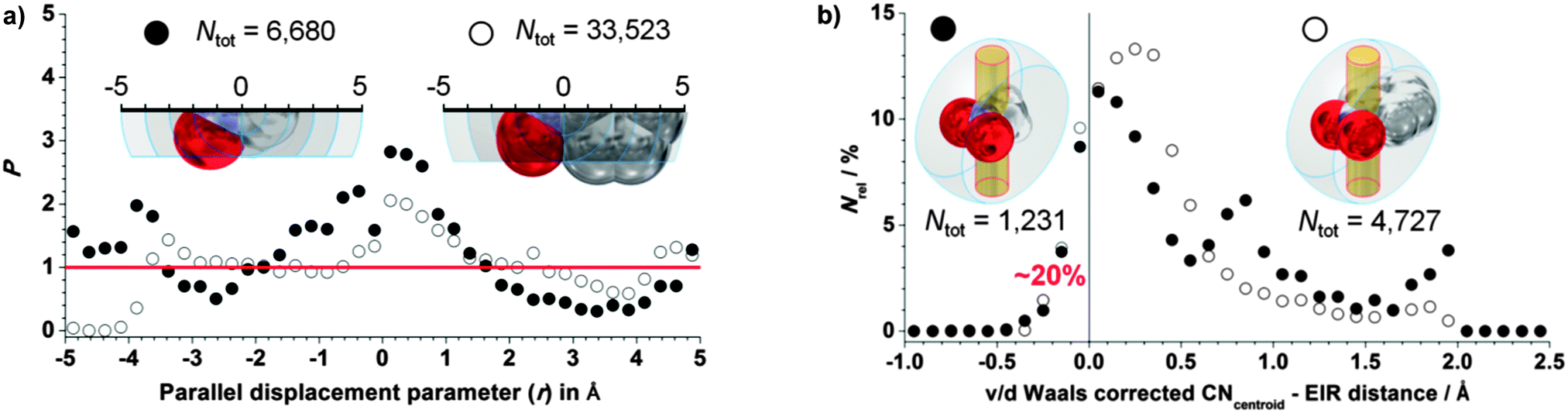 | ||
| Fig. 3 Analysis of the directional nature of CNO2 π-holes. Left: directionality plots of P vs. r for CH3NO2⋯ElR (●, Ntot = 6680) and C6H2NO2⋯ElR (○, Ntot = 33523) of the data characterized by D ≤ 5 Å and y ± 2 Å; right: Nrelativevs. v/d Waals corrected CNcentroid–ELR distance (assuming (vdWC + vdWN)/2 = 1.625 Å) for CH3NO2⋯ElR (●, Ntot = 1231) and C6H2NO2⋯ElR (○, Ntot = 4727) of the data characterized by D ≤ 5 Å and r ± 1 Å. | ||
We have theoretically analysed (see the ESI† for details) several representative complexes between electron-rich moieties and nitrobenzene (Fig. 4a; a similar study with nitromethane is reported elsewhere).11 The interaction energies and some geometric features of the complexes are summarized in Table 1. The interaction energies are similar for nearly all complexes ranging from −2.3 to −6.6 kcal mol−1 and the most favourable one corresponds to chloride complex 7. The π-hole in nitrobenzene is located along the C–N bond and close to the nitrogen atom (see Fig. 1f and 4b). We have included both ElR⋯N and ElR⋯C distances (denoted as DN and DC, respectively) in Table 1. All complexes exhibit similar DN and DC distances and in all cases the electron-rich atom is located over the C–N bond, in agreement with results of the MEP analysis. The interaction energy is modest in complex 6 due to the low basicity of the sp-hybridized nitrogen atom of the HCN molecule.
 | ||
| Fig. 4 (a) Complexes 1–7 studied in this work. (b) A pictorial representation of the complementarity of the MEP surfaces of nitrobenzene and trimethylamine. | ||
| Complex | E | D N | D C |
|---|---|---|---|
| 1 | −6.2 | 2.770 | 2.932 |
| 2 | −4.3 | 3.636 | 3.547 |
| 3 | −4.4 | 2.825 | 2.931 |
| 4 | −4.7 | 3.416 | 3.448 |
| 5 | −4.7 | 2.956 | 3.001 |
| 6 | −2.3 | 3.181 | 3.191 |
| 7 | −6.6 | 3.204 | 3.047 |
The optimized geometries of some complexes are shown in Fig. 5, including the “atoms-in-molecules”16 (AIM) distribution of critical points computed for complexes 1 (lone pair donor ElR) and 5 (anionic ElR). The optimized geometries and critical point distribution for the rest of the complexes are included in the ESI.† It can be clearly observed in Fig. 5 and Fig. S1 (ESI†) that the position of the ElR atom of the interacting molecule coincides with the location of the π-hole in all complexes, indicating that the interaction is basically electrostatic in nature. The AIM analysis of 1 is complicated since it shows five bond critical points symmetrically distributed. One bond critical point (ρ = 1.54 × 102 a.u.) connects both nitrogen atoms characterizing and confirming the π-hole interaction. In addition, two bond critical points (ρ = 0.62 × 102 a.u.), which connect two hydrogen atoms of trimethylamine to two carbon atoms of the ring, reveal the presence of two C–H/π interactions. Moreover, two additional bond critical points (ρ = 0.70 × 102 a.u.) connecting two hydrogen atoms to the oxygen atoms of the nitro group, reveal the presence of two C–H⋯O interactions. The charge density computed at the bond critical point that connects both nitrogen atoms is considerably greater than the ones measured at the other bond critical points (1.54 × 102vs. ∼0.66 × 102). Therefore, the complexation is dominated by the π-hole interaction with some contribution of the other two interactions. In complex 7, the distribution shows a single bond critical point that connects Cl− to the carbon atom, instead of the nitrogen atom, confirming the interaction.
 | ||
| Fig. 5 (a–c) Optimized π-hole complexes 1, 3 and 7 studied in this work. (d and e) Distribution of critical points in complexes 1 and 7 (bond and ring critical points are represented by red and yellow spheres, respectively). Bond paths connecting bond critical points are represented. | ||
In conclusion, we have demonstrated that the nitro groups in nitromethane and nitrobenzene are able to interact favourably with electron-rich molecules by means of the π-hole that is located above and below the C–N bond. The interaction is characterized by the presence of a bond critical point that connects the ElR atom to the C–N bond. More importantly, the statistical survey of the CSD study reveals that the interaction between the π-holes of nitro-groups and electron-rich atoms is directional. Nitro groups are widespread amongst small organic molecules and to date the protein data bank (PDB) contains 380 ligands that bear an –NO2 moiety. It is thus reasonable to anticipate an experimental study unveiling the functional relevance of this novel π-hole interaction.
AB and AF thank MINECO of Spain (projects CTQ2011-27512/BQU and CSD2010-00065, FEDER funds) for financial support.
Notes and references
- (a) H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed; (b) H. J. Schneider and A. Yatsimirski, Principles and methods in supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (b) M. Brandl, M. S. Weiss, A. Jabs, J. Suhnel and R. Hilgenfeld, J. Mol. Biol., 2001, 307, 357–377 CrossRef CAS PubMed.
- (a) Hydrogen Bonding: New Insights, ed. S. Grabowski, Springer, Heidelberg, 2006 Search PubMed; (b) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC; (c) E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC.
- (a) M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613–10620 CrossRef CAS PubMed; (b) D. B. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638–10639 CrossRef CAS PubMed; (c) P. Sanz, O. Mó and M. Yáñez, Phys. Chem. Chem. Phys., 2003, 5, 2942–2947 RSC.
- (a) S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem. – Eur. J., 2011, 17, 6034–6038 CrossRef CAS PubMed; (b) S. Scheiner, J. Chem. Phys., 2011, 134, 094315 CrossRef PubMed; (c) J. E. Del Bene, I. Alkorta, G. Sánchez-Sanz and J. Elguero, J. Phys. Chem. A, 2012, 116, 9205–9213 CrossRef CAS PubMed; (d) P. Kilian, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J., 2003, 9, 215–222 CrossRef CAS PubMed.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed; (b) S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824–1834 RSC; (c) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. – Eur. J., 2014, 20, 10245–10248 CrossRef PubMed; (d) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626–12629 RSC.
- (a) H. B. Burgi, Inorg. Chem., 1973, 12, 2321–2325 CrossRef; (b) H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS; (c) H. B. Burgi, J. D. Dunitz, J. M. Lehn and G. Wipff, Tetrahedron, 1974, 30, 1563–1572 CrossRef.
- (a) P. H. Maccallum, R. Poet and E. J. Milnerwhite, J. Mol. Biol., 1995, 248, 374–384 CAS; (b) G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615–620 CrossRef CAS PubMed; (c) M. Harder, B. Kuhn and F. Diederich, ChemMedChem, 2013, 8, 397–404 CrossRef CAS PubMed.
- (a) T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 3902–3906 RSC; (b) T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 1027–1030 RSC.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed.
- A. Bauza, R. Ramis and A. Frontera, J. Phys. Chem. A, 2014, 118, 2827–2834 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- K. E. Ranaghan, J. E. Hung, G. J. Bartlett, T. J. Mooibroek, J. N. Harvey, D. N. Woolfson, W. A. van der Donk and A. Mulholland, Chem. Sci., 2014, 5, 2191–2199 RSC.
- (a) T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 1802–1805 RSC; (b) T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 4565–4570 RSC.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 8462–8467 RSC.
- (a) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, USA, 1994 Search PubMed; (b) R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS; (c) R. F. W. Bader, Atoms in Molecules, Encyclopaedia of Computational Chemistry, 1998, vol. 1, pp. 64–86 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc09132a |
| ‡ Nr = the number of hits in between two r-values; Ntotal = the total number of hits; Vfreer = the volume in between two r-values minus the volume of the host in between these two r-values (hence, the free volume), as found within a 4 Å wide symmetrical spherical segment of a sphere with a 5 Å radius; Vfreetotal is the volume of a 4 Å wide symmetrical spherical segment of a sphere with a 5 Å radius minus the volume of the host in that body. Models of the central groups were constructed using averages of interatomic distances and angles and assuming generally accepted van der Waals radii. All volumes were computed using Autodesk® Inventor® Professional. |

| This journal is © The Royal Society of Chemistry 2015 |