
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid ultrasonic isothermal amplification of DNA with multiplexed melting analysis – applications in the clinical diagnosis of sexually transmitted diseases†
Gaolian
Xu
a,
Rory N.
Gunson
b,
Jonathan M.
Cooper
a and
Julien
Reboud
 *a
*a
aDivision of Biomedical Engineering, School of Engineering, University of Glasgow, Oakfield Avenue, Rankine Building, G12 8LT Glasgow, UK. E-mail: julien.reboud@glasgow.ac.uk
bConsultant Clinical Scientist, West of Scotland Specialist Virology Centre, Glasgow Royal Infirmary, G31 2ER, UK
First published on 23rd December 2014
Abstract
We describe a nucleic acid testing (NAT) platform for infectious disease diagnostics at the point-of-care, using surface acoustic waves (SAW) to perform a multiplexed loop-mediated isothermal amplification (LAMP) test for sexually transmitted diseases. The ultrasonic actuation not only enables faster NAT reactions but also provides a route towards integrating low-cost, low-power molecular diagnostics into disposable sensors.
Infectious diseases cause more than half of the deaths in low resource countries.1 To reduce the impact of these diseases, it is now broadly accepted that early diagnosis is needed in order to break the cycle of infection and transmission, reducing both mortality and morbidity. The development of rapid, high performance molecular diagnostic technologies, such as those involved in NATs has the potential to provide a much-needed step change, enabling the early diagnosis of infection.2
Currently NATs can provide information on the microbial species and sub-types, enabling, for example, the identification of drug resistant strains. However, existing technologies suffer from limitations in both speed and cost.2 They also often require considerable user expertise. To date the majority of these have been based upon the polymerase chain reaction (PCR), an amplification method which although widely used, is also difficult to implement in resource limited areas.2 As an alternative loop mediated isothermal amplification (LAMP) can amplify DNA at a constant temperature (60–65 °C), enabling detection with both high sensitivity and specificity.3 The technique has previously been demonstrated for pathogen detection, such as Mycobacterium tuberculosis4 and HIV,5 and has led to its adoption by important stakeholders such as FIND.6
Recently, LAMP amplification has also been integrated within microfluidic devices to enable small volume analysis and enhance portability.7 Real-time sensing has also been performed using both fluorescence3 and turbidity (which can be seen with the naked eye, but does not enable multiplexing).8
Surface acoustic waves (SAW) are most commonly generated with an interdigitated transducer (IDT) patterned on the surface of a piezoelectric substrate. The energy in the associated ultrasonic wave is located at the chip surface, in this case at its interface with the sample, positioned in the propagation pathway (Fig. 1).9 Upon reaching the liquid, the SAW refracts and depending upon the power and frequency, the energy can be used for a range of microfluidic functions, including heating and mixing.10 We have previously shown that SAWs can be coupled into a disposable chip and then used to perform sample processing of blood,11 to control aggregation of colloids,12 to concentrate micro-organisms in blood13 and integrate PCR assays onto a low-cost low-power system.14
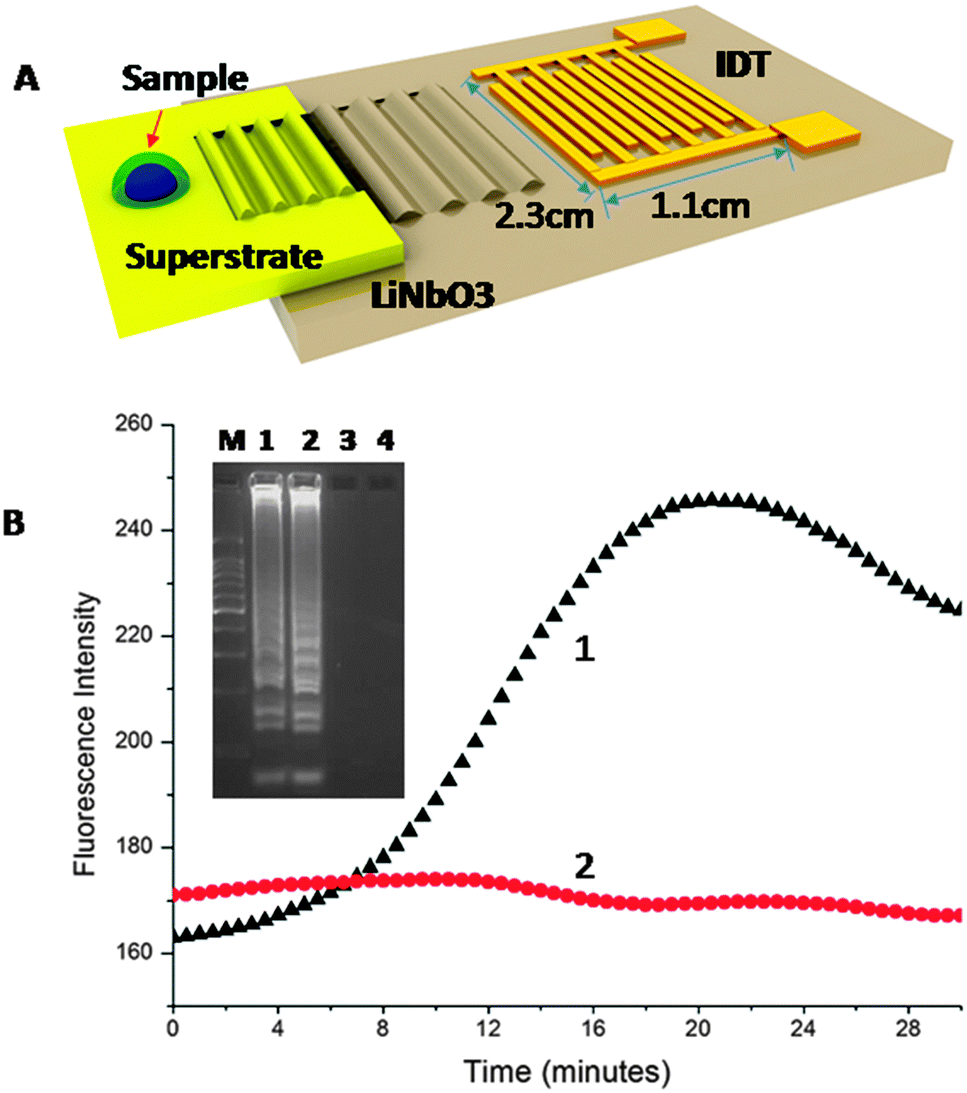 | ||
| Fig. 1 (A) Integrated SAW-LAMP device. The SAW generated by the IDT patterned on the piezoelectric surface was coupled into a superstrate. A 4.0 μL droplet of the LAMP mix was dispensed on the superstrate and covered with mineral oil to prevent evaporation. (B) Real time results of SAW-LAMP: (1) black triangle, positive amplification (103 copies per reaction); (2) red circles, negative amplification (non-template signal). The target gives an amplification after ∼8 min, while the negative sample shows a constant signal. (inset) Electropherograms of SAW-LAMP of CT: Lane M: DNA marker (100 bp ladder – Promega G2101); Lane 1: SAW-LAMP positive; Lane 2: LAMP (positive) with thermal heating actuation using a Peltier heater; Lane 3: SAW-LAMP non-template negative control; Lane 4: LAMP non-template negative with thermal heating actuated using a Peltier heater. | ||
In this paper, we demonstrate the integration of SAW-induced heating to perform real-time LAMP using EvaGreen™ fluorescent DNA-intercalating dye system.15 We first characterised the sensitivity of the technique for the detection of Chlamydia trachomatis (CT), demonstrating limits of detection of ten copies of the CT target per sample (established using a serial dilution). We also showed for the first time that SAW-induced amplification provides a new method to enable multiplexed detection, using melting curve analysis to simultaneously detect Neisseria gonorrhoea (GC) and CT. The LAMP reaction was performed at a constant temperature, without the requirement for precise control of thermal cycles. As a consequence, we demonstrated an integrated assay protocol with greatly reduced reaction times and a simplified operational control. Interestingly, the use of acoustic actuation results in streaming flows that speed up the LAMP reaction, when compared to electrical heating, by as much as 20%.
The assay was developed on a disposable chip by in-coupling the SAWs propagated at 20 MHz (±3.0 MHz), from a LiNbO3 piezoelectric substrate. The sample was a 4.0 μL drop of LAMP mix (see supplementary methods in ESI† for details on the reagents and DNA sequence designs), encapsulated in mineral oil to prevent evaporation (Fig. 1A). Heat was generated using the SAW and temperature changes were monitored, for purposes of characterisation and calibration using infrared imaging (Fig. S1 in ESI†). An actuation power (ca. −1.2 W) was used to stabilise the temperature of the sample at 60 °C (Fig. S1 in ESI† – temperature stability). As amplification took place, the EvaGreen™ fluorescent dye was incorporated into the double-stranded DNA, with a resulting exponential increase in the signal intensity, Fig. 1B.
The fluorescence intensity of the non-template negative control (no target) remained constant throughout the assay, indicative of no non-specific amplification (Fig. 1B). After 20 min, the efficiency of the fluorescent dye diminished, resulting in a decrease in the intensity of the signal. Gel electrophoresis of the amplified target confirmed a positive amplification using both SAW (Lane 1) and Peltier actuated heating (Lane 2) with no non-specific signal for the negative samples (Lanes 3 and 4), Fig. 1B (inset). We further showed the specificity of the reaction and its applicability to clinical diagnostics by processing two residual GC positive patient samples (obtained as swabs and extracted as part of routine clinical diagnostics at the NHS West of Scotland Specialist Virology Centre – Fig. S3 in ESI†). The analysis of the samples demonstrate the specificity of the CT SAW LAMP assay, as no non-specific signal can be seen.
To provide a figure of merit for the sensitivity, we serially diluted the CT DNA target (the cryptic plasmid gene of CT,16 using the primer set of Table S1 in ESI†) from 105 to 10 copies per reaction. Fig. 2A shows normalised real-time amplification curves for each concentration. As the copy number was decreased, the exponential phase of signal enhancement started later as a consequence of the reaction kinetic (from ca. 6 min for 105 copies to ca. 22 min for 10 copies, Fig. 2A).
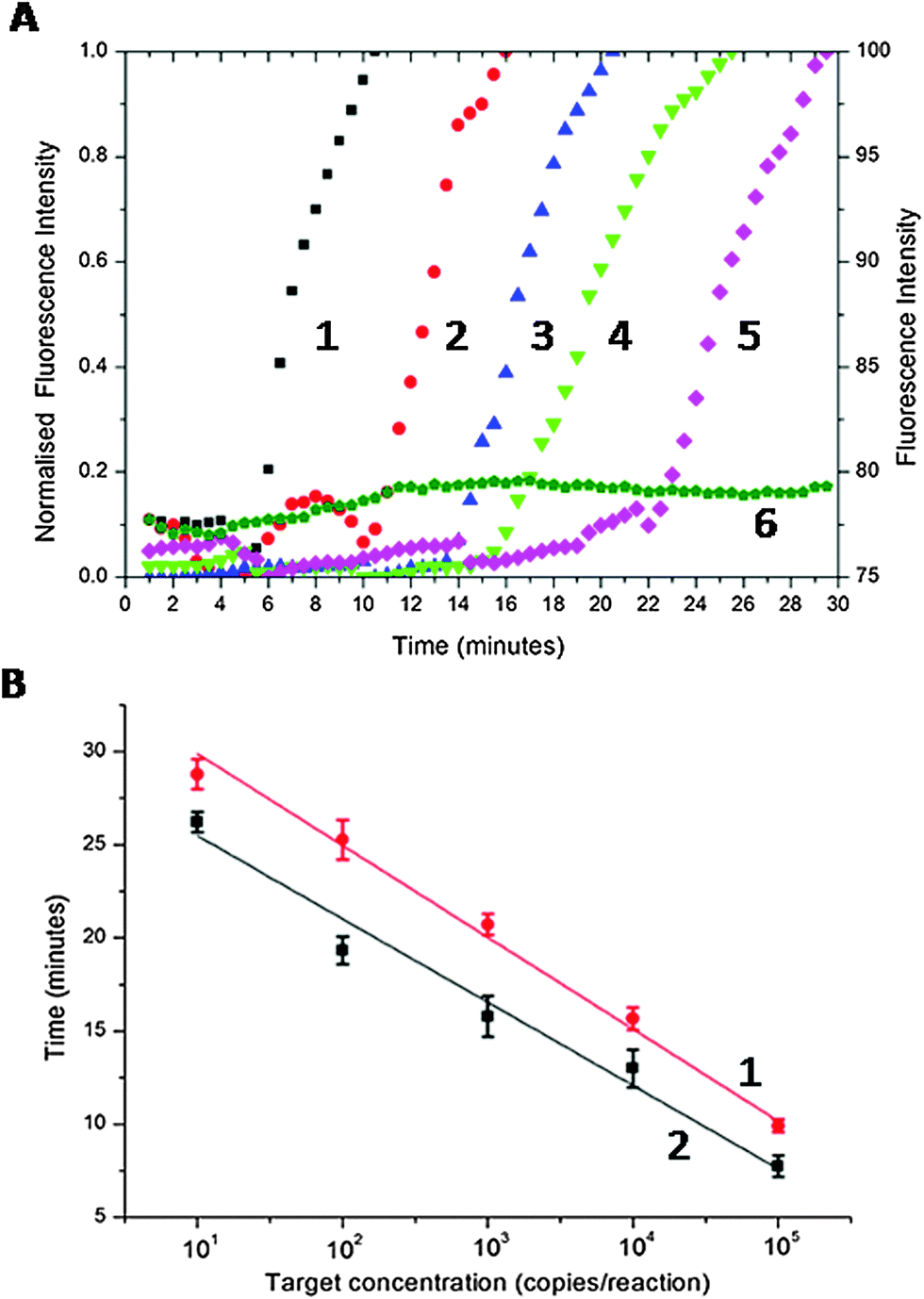 | ||
| Fig. 2 (A) Real-time amplification curve of SAW LAMP of serial 10-fold diluted CT positive template (1–5 normalised real-time amplification curves, left axis), and ddH2O as a negative control (6, right axis, not normalised to ease readability): (1) 105 copies per reaction (black square); (2) 104 copies per reaction (red circle); (3) 103 copies per reaction (blue up-triangle); (4) 102 copies per reaction (green triangle); (5) 10 copies per reaction (magenta diamond). (6) Negative control (olive pentagon). As the concentration increases, the amplification is initiated earlier, evidenced by the exponential increase in the fluorescence. (B) Threshold time (defined as the time corresponding to 50% of the maximum fluorescence intensity, Tt) as a function of target concentration, (1) Peltier-based LAMP (red disks) and (2) SAW LAMP (dark square). Data is the average of at least 3 replicates and error bars represent the standard deviation. The data was fitted with linear regression (R2 > 0.98). | ||
To demonstrate the efficiency and speed of the SAW-actuated assay, we defined a threshold time (Tt) as the reaction time for the fluorescence signal to reach 50% of the maximum (this new figure of merit is analogous to the cycle threshold (Ct) of real-time PCR).17Fig. 2B shows that, as the target concentration was increased, Tt decreases linearly with the log of the target concentration.18 The precise nature of this relationship is important as it enables us to quantify the amount of target DNA present in the sample, prior to LAMP amplification. The divergent gradients of the linear regressions in Fig. 2B indicate the relative difference in speed of the SAW and the Peltier-based assays. SAW induced streaming enhanced the speed of response (SAW-based LAMP were on average 18.2% ± 2.5 faster than the Peltier system, Fig. 2B) providing a clear analytical advantage for assays that require higher sensitivities. It is particularly relevant to note that the response time of a point-of-care diagnostic assay and consequently the time that patients wait for a result is critical in compliance in both screening and point-of-care testing.19 This is especially relevant for sexually transmitted infection (STI) testing, where detecting low copy numbers quickly, is critical to avoid complications, especially in asymptomatic infected (sub-clinical) patients, by enabling early diagnosis and treatment.20
GC and CT are two of the most common bacterial STIs,20 and a multiplexed assay would therefore not only provide an improved clinical outcome for the individual (as patients are often co-infected with both pathogens), but would also reduce the overall levels of the diseases in the population. To enable multiplexing, we used the amplicons' different melting temperatures to distinguish between the two diseases. We first established the specificity of each singleplex reaction, using melting analysis, for each pathogen, whilst ramping the temperature between 60 °C and 90 °C (Fig. 3A). The specific melting temperature was defined as the inflexion point at which dF/dT reaches a minimum (where F is the fluorescence intensity and T is temperature), Fig. 3C. The melting temperature of CT amplicons obtained using SAW actuation was 79.65 ± 0.14 °C, whilst that for GC was 83.55 ± 0.53 °C (Fig. 3B). An example of differentiated curves is available as Fig. S2 in ESI.†
 | ||
| Fig. 3 LAMP product melting analysis using EvaGreen™. (A) Shows the melting curves of CT and GC as singleplexes. As the temperature was increased from 60 °C to 90 °C, the double stranded DNA became single stranded, causing the decrease of fluorescence intensity. The number 1–4 in A1/A2 show the different targets: (1) CT positive (103 copies per reactions); (2) GC positive (103 copies per reactions); (3) GC negative (ddH2O); (4) CT negative (ddH2O). (B) Shows the melting temperature of CT and GC. Data is the average of a minimum of 3 repeats and error bars represent the standard deviation. (C) CT/GC multiplex reaction with different target. (C) Shows the relationship between dF/dT and temperature, where F is the fluorescence intensity and T is the temperature. (1) Negative (ddH2O); (2) GC positive only (103 copies per reactions); (3) GC/CT positive (103 copies per reactions); (4) CT positive only (103 copies per reactions). | ||
By combining amplicons from both target pathogens in the same sample, we were able to use the melting curves to provide a multiplexed SAW LAMP assay, Fig. 3C. The difference between the melting temperatures for each pathogen was sufficiently large to distinguish between the targets (Student's T-test P > 0.95), enabling the diagnosis of both diseases simultaneously. This technique is currently limited by the availability of amplicons with significantly different melting temperatures.21 In future, multiplexing could be further extended through the use of more complex strategies such as high resolution melting (HRM).22 Despite challenges in combining a high number of primers in a single reaction (6 per target), previously it has been shown that specific strategies can enabled the multiplexing of up to four different targets in one sample.23
In conclusion, we show for the first time that acoustic actuation using SAW can be used to perform highly sensitive and specific multiplexed LAMP-based amplification of pathogen DNA (with a limit of detection down to 10 copies) with enhanced speeds. The strategy for multiplexing enables single colour detection, thereby simplifying the control and design of the instrumentation. This ease of implementation, coupled with the use of low-power, mass manufacturable SAW devices,14 provides the potential to significantly impact upon near patient diagnostics.
This work was supported by a College of Science and Engineering Studentship (GX, Glasgow, UK), a Lord Kelvin and Adam Smith Research Fellowship (JR, Glasgow, UK), an EPSRC fellowship (JC, EP/K027611/1), an ERC Advanced Investigator Award (JC), and an NHS Partnership award (JC, JR, RG). The authors thank the James Watt Nanofabrication Centre (Glasgow, UK) for help in device fabrication and David Paterson (Glasgow, UK) for illustration.
Notes and references
- D. M. Morens, G. K. Folkers and A. S. Fauci, Nature, 2010, 463, 122 CrossRef CAS.
- R. McNerney and P. Daley, Nat. Rev. Microbiol., 2011, 9, 204–213 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63–e63 CrossRef CAS PubMed.
- T. Iwamoto, T. Sonobe and K. Hayashi, J. Clin. Microbiol., 2003, 41, 2616–2622 CrossRef CAS.
- N. Hosaka, N. Ndembi, A. Ishizaki, S. Kageyama, K. Numazaki and H. Ichimura, J. Virol. Methods, 2009, 157, 195–199 CrossRef CAS PubMed.
- N. Zablon Kithinji, PLoS Neglected Trop. Dis., 2012, 6, e1572 Search PubMed.
- K. Hsieh, A. S. Patterson, B. S. Ferguson, K. W. Plaxco and H. T. Soh, Angew. Chem., Int. Ed., 2012, 51, 4896–4900 CrossRef CAS PubMed.
- Y. Mori, M. Kitao, N. Tomita and T. Notomi, J. Biochem. Biophys. Methods, 2004, 59, 145–157 CrossRef CAS PubMed.
- A. Wixforth, Spec. Issue Dedic. Profr. Jorg Kotthaus Occas. His 60th Birthd. 29th May 2004, 2003, 33, 389–396.
- X. Ding, P. Li, S.-C. S. Lin, Z. S. Stratton, N. Nama, F. Guo, D. Slotcavage, X. Mao, J. Shi, F. Costanzo and T. J. Huang, Lab Chip, 2013, 13, 3626–3649 RSC.
- J. Reboud, Y. Bourquin, R. Wilson, G. S. Pall, M. Jiwaji, A. R. Pitt, A. Graham, A. P. Waters and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15162–15167 CrossRef CAS PubMed.
- J. Reboud, C. Auchinvole, C. D. Syme, R. Wilson and J. M. Cooper, Chem. Commun., 2013, 49, 2918–2920 RSC.
- Y. Bourquin, A. Syed, J. Reboud, L. C. Ranford-Cartwright, M. P. Barrett and J. M. Cooper, Angew. Chem., Int. Ed., 2014, 53, 5587–5590 CrossRef CAS PubMed.
- Z. Guttenberg, H. Muller, H. Habermuller, A. Geisbauer, J. Pipper, J. Felbel, M. Kielpinski, J. Scriba and A. Wixforth, Lab Chip, 2005, 5, 308–317 RSC.
- Y.-M. Qiao, Y.-C. Guo, X.-E. Zhang, Y.-F. Zhou, Z.-P. Zhang, H.-P. Wei, R.-F. Yang and D.-B. Wang, Biotechnol. Lett., 2007, 29, 1939–1946 CrossRef CAS PubMed.
- K. Jaton, J. Bille and G. Greub, J. Med. Microbiol., 2006, 55, 1667–1674 CrossRef CAS PubMed.
- R. Higuchi, C. Fockler, G. Dollinger and R. Watson, Nat. Biotechnol., 1993, 11, 1026–1030 CrossRef CAS PubMed.
- F. Ahmad, G. Seyrig, D. Tourlousse, R. Stedtfeld, J. Tiedje and S. Hashsham, Biomed. Microdevices, 2011, 13, 929–937 CrossRef CAS PubMed.
- G. J. Kost, N. K. Tran, M. Tuntideelert, S. Kulrattanamaneeporn and N. Peungposop, Am. J. Clin. Pathol., 2006, 126, 513–520 CrossRef PubMed.
- S. B. Lyss, M. L. Kamb, T. A. Peterman, J. S. Moran, D. R. Newman, G. Bolan, J. M. Douglas, M. Iatesta, C. K. Malotte, J. M. Zenilman, J. Ehret, C. Gaydos and W. J. Newhall, Ann. Intern. Med., 2003, 139, 178–185 CrossRef.
- J. Mahony, S. Chong, D. Bulir, A. Ruyter, K. Mwawasi and D. Waltho, J. Clin. Microbiol., 2013, 51, 2696–2701 CrossRef CAS PubMed.
- D. C. T. Ong, W.-C. Yam, G. K. H. Siu and A. S. G. Lee, J. Clin. Microbiol., 2010, 48, 1047–1054 CrossRef CAS PubMed.
- N. A. Tanner, Y. Zhang and T. C. Evans, BioTechniques, 2012, 52, 81–89 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental methods, Fig. S1–S3 and Table S1. See DOI: 10.1039/c4cc08389j |
| This journal is © The Royal Society of Chemistry 2015 |