
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A temporary-bridge strategy for enantioselective organocatalyzed synthesis of aza-seven-membered rings†
Sébastien
Goudedranche
,
David
Pierrot
,
Thierry
Constantieux
,
Damien
Bonne
* and
Jean
Rodriguez
*
Aix Marseille Université, CNRS, Centrale Marseille, ISM2 UMR 7313, 13397, Marseille, France. E-mail: damien.bonne@univ-amu.fr; jean.rodriguez@univ-amu.fr
First published on 31st October 2014
Abstract
We report the first enantioselective organocatalyzed domino synthesis of azepane moieties. This temporary-bridge strategy is based on a conceptually original annulation of ambident electrophilic and 1,4-bis-nucleophilic α-ketoamides with 1,3-bis-electrophilic enals. The obtained oxygen-bridged azepanes can be selectively transformed into optically active azepanone, azepanol or azepanedione derivatives of high synthetic value.
The design of new efficient enantioselective organocatalyzed domino transformations is a very active field of research especially for synthesizing complex cyclic enantiopure molecules from easily accessible acyclic starting materials in an environmentally friendly way.1 In this context, enantioselective organocascades of five-2 and six-membered cycles3 are well documented, while accessing the parent seven-membered cycles still constitutes a real synthetic challenge4,5 due to the need to overcome a negative entropic factor during the ring closing step.6 As rare examples of success in this field, in the benzoxepinone series, two closely related enantioselective carbene-catalyzed formal [4+3] heterocycloadditions have been proposed (Scheme 1a).7 Additionally, a very recent chiral phosphoric acid (CPA)-catalyzed enantioselective intramolecular Mannich reaction has been known to give access to dibenzo[1,4]diazepine scaffolds (Scheme 1b).8 Hence, an efficient complementary organocatalyzed domino reaction to synthesize challenging medium-sized ring systems in optically active forms is highly desirable and of relevance for synthetic purposes. Herein, we report our new organocatalytic approach to synthesize seven-membered rings based on a temporary-bridge strategy,9 which circumvents difficulties associated with the construction of medium-sized rings by direct cyclization routes (Scheme 1c). The designed domino approach is based on organocatalyzed domino formation of bicyclo[3.2.1]octane derivative 3. The overall cascade is thermodynamically driven by the final bridging step, when properly functionalized 1,4-bis-nucleophile 1 bearing an additional electrophilic center reacts with 1,3-bis-electrophile 2 possessing a latent nucleophile that results in the concomitant formation of three covalent bonds. In a subsequent step, the bridge could be selectively cleaved (revealing step), allowing the formation of a highly functionalized optically active seven-membered ring system 4.
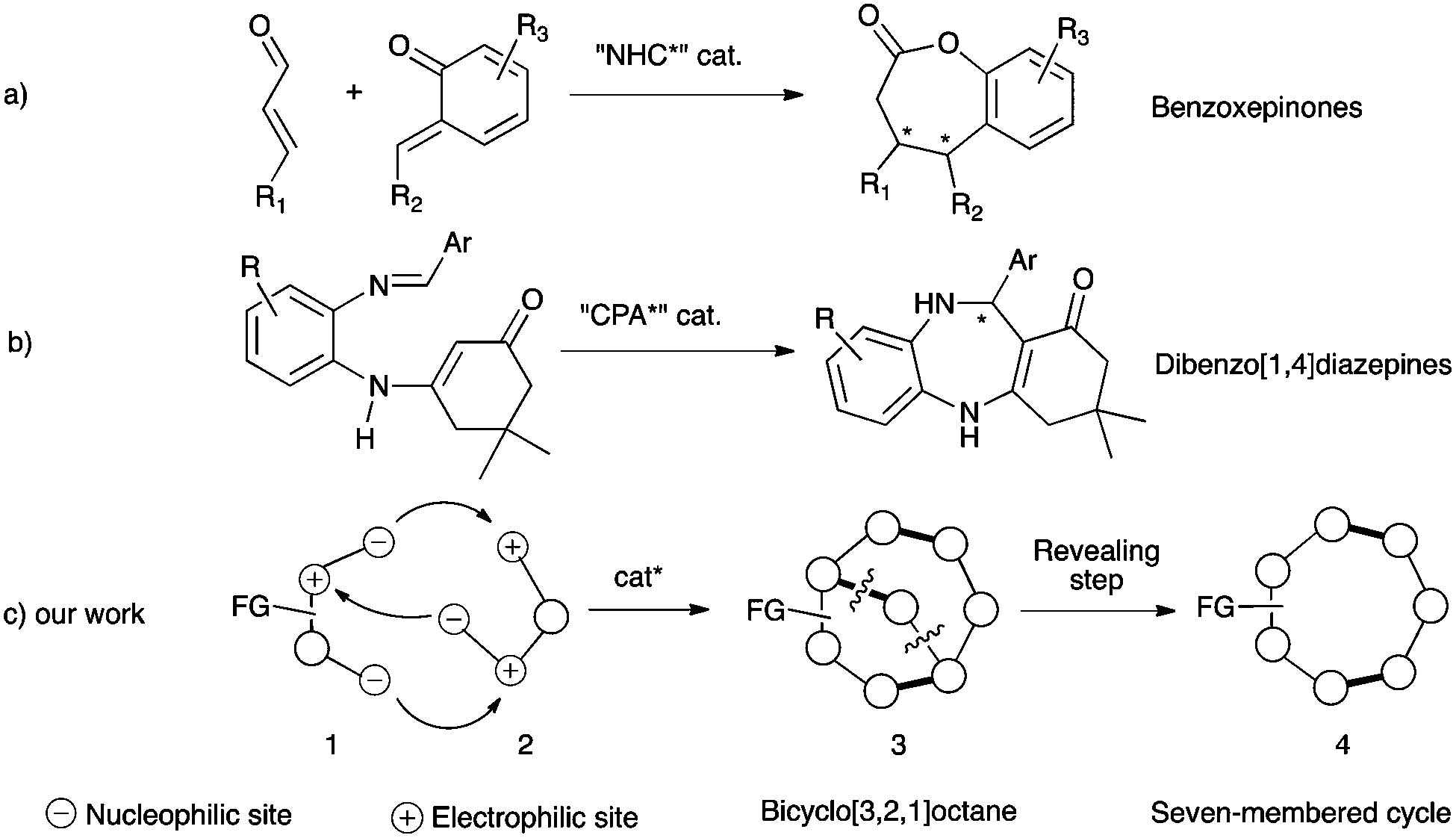 | ||
| Scheme 1 Organocatalytic approaches and the temporary-bridge strategy to synthesize seven-membered cycles. | ||
To pursue this new strategy, the selection of the two bridging units is crucial in terms of ready availability and compatibility with the mild reaction conditions required to reach a high level of stereocontrol. We propose the use of α-ketoamides 1,10 possessing multiple reactive sites,11 as potential 1,4-bis-nucleophiles that could be combined with 1,3-bis-electrophilic enals 2 following a Michael–hemiaminalization sequence affording intermediate 5 (Scheme 2).12 This step would be potentially highly reversible and contra-thermodynamic but the possible temporary formation of an internal oxygen tether by a final intramolecular hemiacetalization step of the highly electrophilic ketone could drive the reaction toward the formation of 3 possibly stabilized by an intramolecular hydrogen bond. The overall thermodynamically driven process allows direct access to a 7-aza-8-oxa-bicyclo[3.2.1]octane with concomitant formation of three covalent bonds and the creation of up to four stereogenic centers. This molecular scaffold is found in several natural products such as the neurotoxin samandarine,13 the antimalarial ribasine14 or deglycocadambine, a new alkaloid recently isolated from the twigs and the leaves of Emmenopterys henryi.15 Moreover, taking advantage of the ring strain release by selective cleavage of the oxa-bridged system will provide a straightforward access to highly functionalized optically active azepane derivatives.
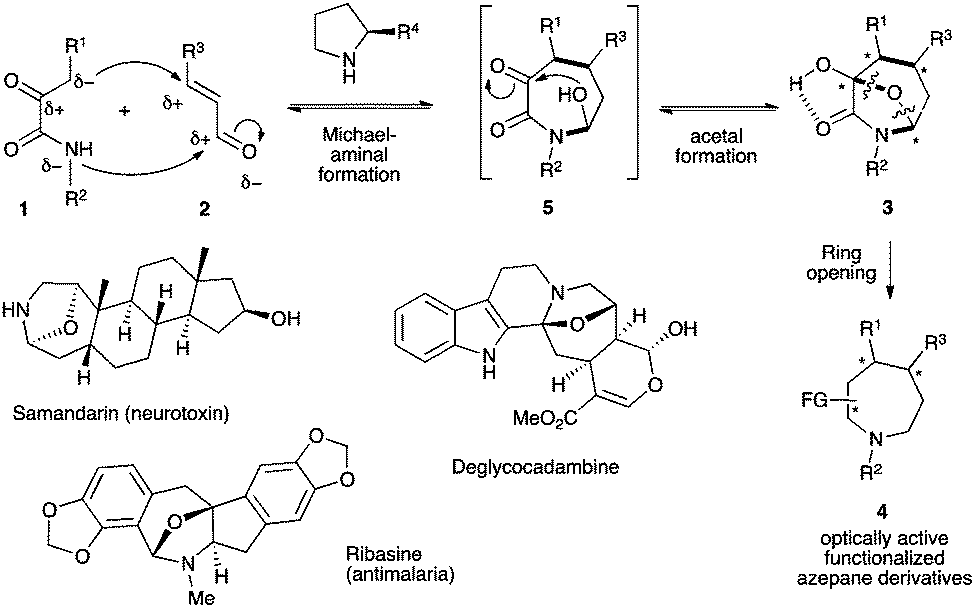 | ||
| Scheme 2 Use of α-ketoamides 1 for the synthesis of nitrogen-containing seven-membered cycles. | ||
We began to test our designed system for the synthesis of bicyclic products 3. We first investigated this reaction with ketoamide 1a and cinnamaldehyde (2a) as starting materials using Hayashi–Jørgensen catalyst 6 (Table 1).16 The reaction conducted in toluene did not proceed at all after 15 h (entry 1). Dichloromethane as well as acetonitrile proved to be unsuitable solvents for this reaction, as only small quantities of Michael adducts were recovered after similar reaction times (entries 2 and 3). Gratifyingly, the use of ethanol afforded the desired product 3a in very good yield, as only two diastereomers (dr = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and with excellent enantioselectivity for both diastereomers (entry 4). Slightly more acidic trichloroethanol allowed the formation of 3a with a better diastereoselectivity while high yield and high enantioselectivity were maintained (entry 5). Finally the reaction temperature was lowered to −7 °C,17 which resulted in an increase of the diastereoselectivity to 8:1 even if a prolonged reaction time was required to drive the reaction to completion (entry 6).
:1) and with excellent enantioselectivity for both diastereomers (entry 4). Slightly more acidic trichloroethanol allowed the formation of 3a with a better diastereoselectivity while high yield and high enantioselectivity were maintained (entry 5). Finally the reaction temperature was lowered to −7 °C,17 which resulted in an increase of the diastereoselectivity to 8:1 even if a prolonged reaction time was required to drive the reaction to completion (entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time | Yieldb (%) | drc | eed,e (%) |
| a Reaction conditions: ketoamide 1a (0.2 mmol, 1.0 equiv.) and catalyst 6 (10 mol%) were dissolved in the solvent (2 mL) and stirred for 15 min (0 °C or −7 °C). Then, cinnamaldehyde (2a) (2 equiv.) was added and the mixture was stirred for the time and temperature indicated. b Isolated yields after flash chromatography. c Determined by 1H NMR of the crude reaction products. d Determined by chiral HPLC analysis. e Values in brackets are for the minor diastereomer. f 10–15% of the Michael adduct was isolated. g The reaction was conducted at −7 °C. | |||||
| 1 | Toluene | 15 h | 0 | — | — |
| 2 | CH2Cl2f | 15 h | 0 | — | — |
| 3 | CH3CNf | 15 h | 0 | — | — |
| 4 | EtOH | 15 h | 92 | 3:1 |
97 [86] |
| 5 | CCl3CH2OH | 24 h | 91 | 5:1 |
99 [88] |
| 6 | CCl3CH2OHg | 3 d | 94 | 7:1 |
99 |
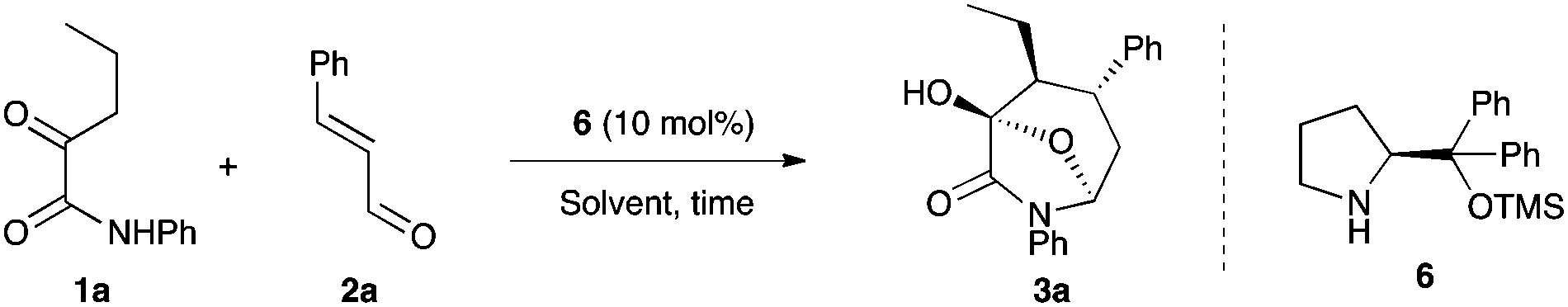
With this final set of optimized conditions in hand,18 the scope of the three bond-forming process was next explored with a variety of 3-aryl α,β-unsaturated aldehydes (Scheme 3). The reaction worked well with aromatic rings bearing either electron-donating or electron-withdrawing groups including sterically demanding ortho functionalization or an anthracenyl substituent, as well as a heteroaromatic ring. The corresponding products 3b–e were obtained in good to high yields with acceptable to very high diastereoselectivities from 4:1 to >20:1 dr, but with uniformly high enantioselectivities in the range 95–97% ee. Similarly, it was found that a broad range of ketoamides 1 could readily participate in this reaction. Increasing the size of the R1 group resulted in generally higher diastereoselectivities (>20:1 dr for R1 = i-Pr in 3c, 3i, 3n, and 3q). Interestingly, the use of 2-oxo-N-phenylpropanamide (1c, R1 = H) resulted in the formation of the desired bicyclic products 3f and 3g as single diastereomers with excellent enantioselectivities (ee = 97%). These results indicate a highly diastereoselective intramolecular hemiacetalization with the stereoselective creation of two stereogenic centers, which is consistent with the fact that only two diastereomers were produced when α-ketoamides with R1 not equal to H were involved.
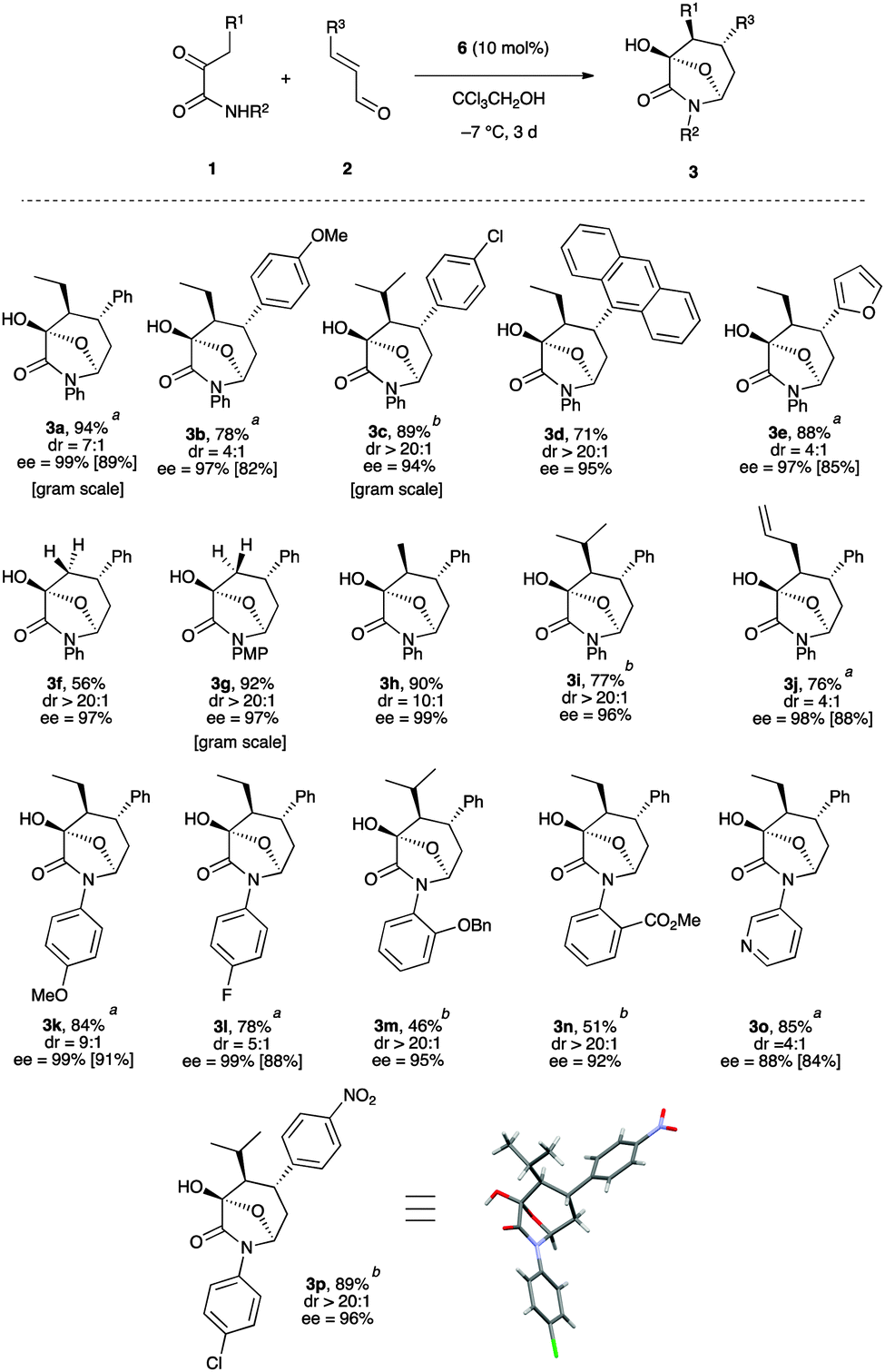 | ||
| Scheme 3 Scope of the domino temporary-bridge approach. a Values in brackets are for the minor diastereomer. b The reaction was conducted at 0 °C. | ||
In addition, α-ketoamides having various electron-donating or electron-withdrawing substituents on the phenyl ring or bearing a pyridine nucleus also proceeded very well in this cascade reaction, thus affording the azepane derivatives (3k–p) in good to excellent yields and high diastereo- and enantioselectivities.19 Finally, the robustness of the transformation was shown by performing gram scale preparations of 3a, 3c and 3g, which proceeded without any difficulty in excellent yields with good stereoselectivities. Concerning the stereochemical assignment, 3p was crystallized and both the relative and absolute configurations of these bicyclic structures were unambiguously identified by X-ray diffraction as (3R,4S,5R,7S).20
Scheme 4 shows our current understanding of the reaction pathway and its thermodynamically driven stereochemical outcome. The initial kinetically controlled Michael addition step involves (Z)-enolate 7 and iminium ion 8.21 In the resulting conformationally restrained environment, the Re face of the thermodynamic (Z)-enolate 7 preferentially interacts with the Si face of iminium ion 8 accounting for the formation of the (3S,4R)-Michael adduct 9. Then, nucleophilic addition of the amide nitrogen atom to give azepane 5 could in theory occur on both diastereotopic faces of aldehyde 7 giving rise reversibly to the two resulting diastereomers 3 and 3′ after an hemiacetalization step. However, the formation of bicyclic product 3′ is hampered by the presence of strong 1,3-diaxial steric interactions between the R2 substituent and the amide moiety.
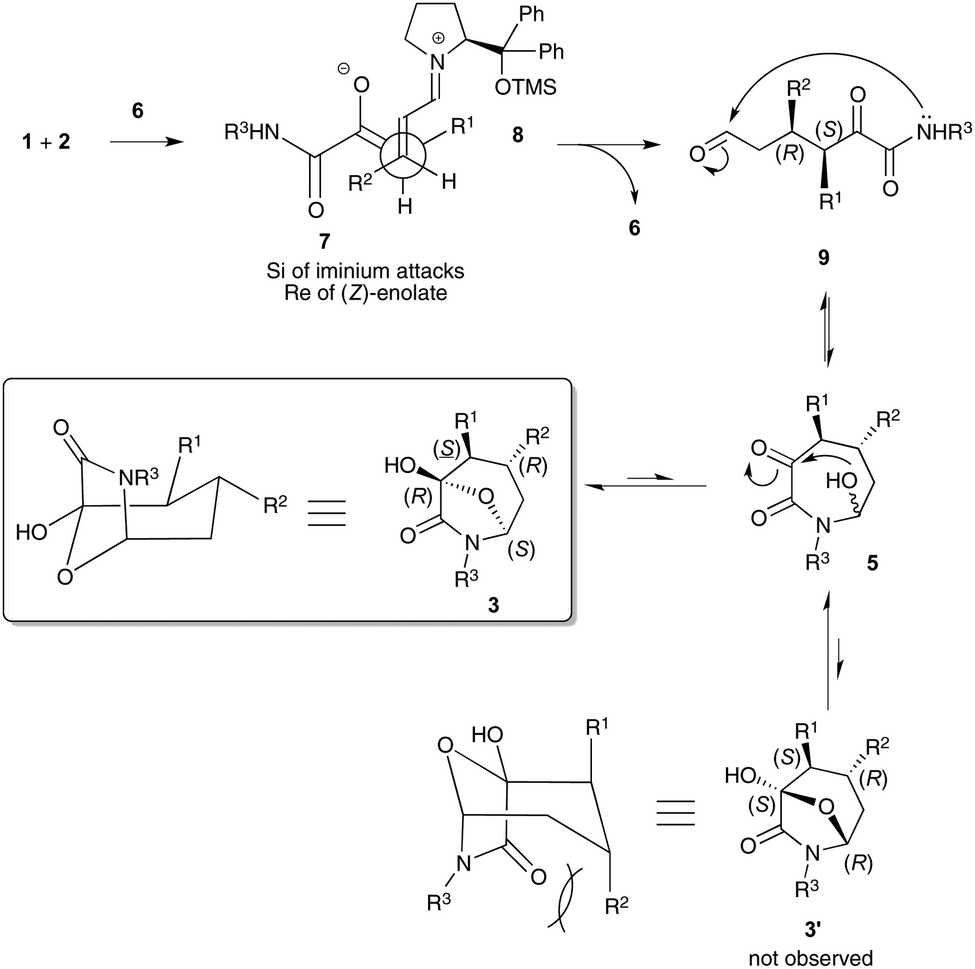 | ||
| Scheme 4 Plausible thermodynamically driven mechanism. | ||
Following our initial temporary-bridge strategy to synthesize azepane rings, we next moved to the development of synthetically useful transformations of bicyclic products 3 in order to release highly functionalized optically active azepanes. These heterocycles are present in numerous natural products and biologically interesting molecules such as (−)-balanol,22 (−)-cobactin T,23 and SQ-33351 (Fig. 1).24 Therefore, a general and efficient strategy to afford such azacyclic rings would be very useful to design new synthetic routes toward these bioactive compounds and their analogs.
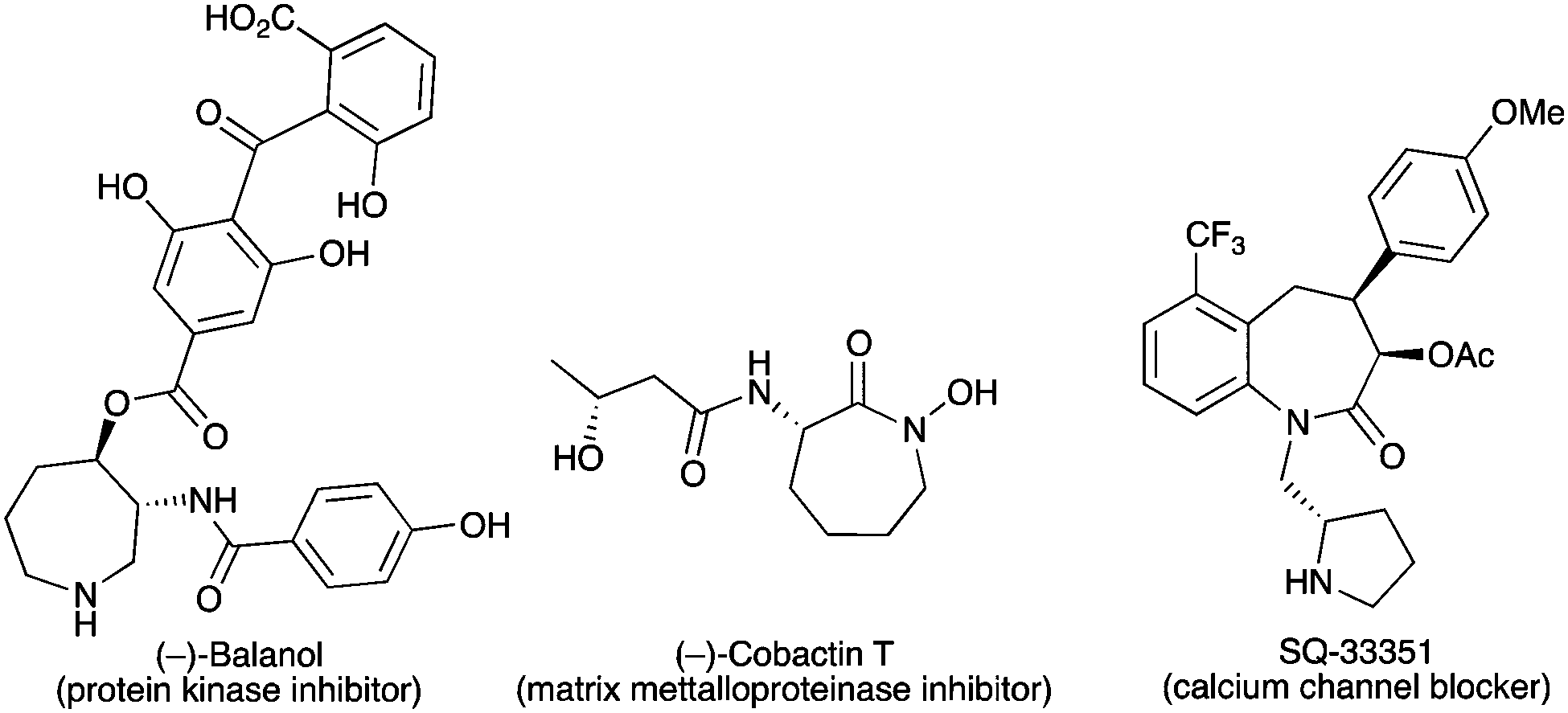 | ||
| Fig. 1 Examples of natural and non-natural products featuring the chiral azepane scaffold. | ||
First, bicyclic ring 3c was easily reduced using the borane–THF complex (1.05 equiv.) to afford azepan-3-one moiety 4a in good yield with no erosion of chiral information (Scheme 5). Also, using three equivalents of the BH3–THF complex, the ketone moiety could be diastereoselectively reduced yielding azepan-3-ol derivative 4b bearing three contiguous stereogenic centers in good yield and excellent stereoselectivity on a synthetically useful mmol scale. This transformation was accomplished on other bicyclic substrates 3 with similar success (see ESI†). Next, we found that Martin's sulfurane reagent25 efficiently triggered a remote dehydration of 3c giving the corresponding unsaturated azepan-2,3-dione 4c in good yield with full retention of the stereochemical information. Interestingly, adding two equivalents of 1,1,1,3,3,3-hexafluoro-2-phenylpropan-2-ol in the reaction mixture furnished the functionalized azepan-2,3-dione 4d bearing three stereogenic centers with an interesting 15:1 dr and a preserved 94% ee. This opens the way for the introduction of an additional point of diversity in the final azepane structure.26
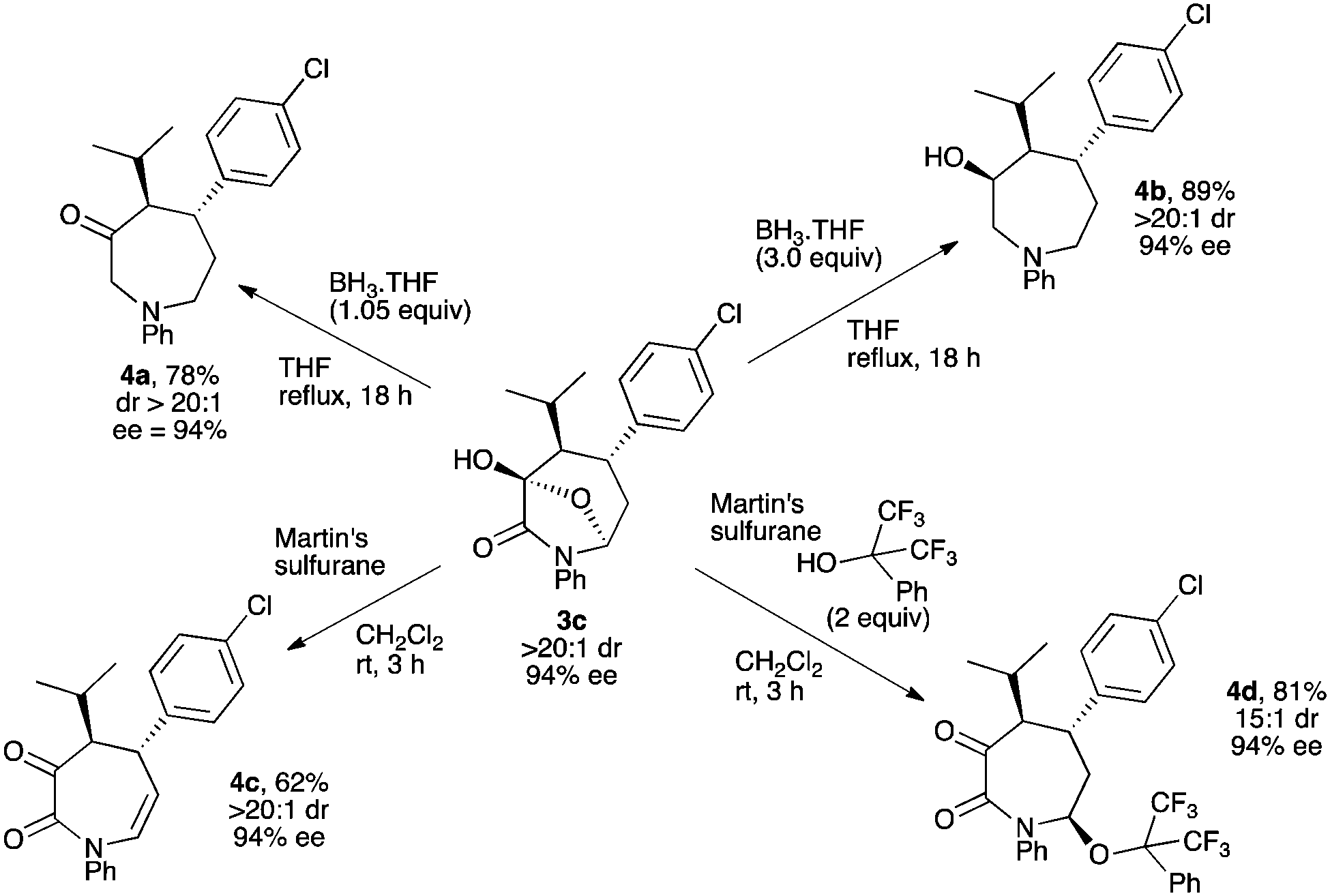 | ||
| Scheme 5 Transformations of bicyclic compound 3c into various functionalized azepanes. | ||
We have demonstrated the feasibility and the efficiency of a new organocatalytic temporary-bridge approach to synthesize optically active nitrogen-containing seven-membered cycles. During this cascade, the synthetic power of α-ketoamides is fully exploited as three complementary reactive sites are involved, triggering an unprecedented thermodynamically driven formal [4+3] heterocyclization. Three chemical bonds (C–C, C–N and C–O) are assembled concomitantly and up to four stereogenic centers are created with very high stereoselectivity. Moreover this domino sequence represents a rare example of an organocatalyzed access to seven-membered ring systems in optically active forms and the first example in the azepanes series. These bicyclic molecules are then easily converted to synthetically valuable azepanone, azepanol or azepanedione derivatives by selective reduction or dehydration providing a complementary answer to the challenging synthesis of optically active seven-membered cyclic nitrogen-containing compounds.
The Agence Nationale pour la Recherche (ANR-11-BS07-0014), the Direction Générale de l'Armement (DGA), the Région PACA, the Centre National de la Recherche Scientifique (CNRS) and the Aix-Marseille Université are gratefully acknowledged for financial support. We also thank Dr N. Vanthuyne and M. Jean (ee measurements), and Dr M. Giorgi (X-ray diffraction analysis).
Notes and references
- For reviews on enantioselective organocascades, see: (a) C. M. R. Volla, L. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed; (b) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed; (c) C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876 CrossRef CAS PubMed; (d) A.-N. Alba, X. Companyo, M. Viciano and R. Rios, Curr. Org. Chem., 2009, 13, 1432 CrossRef CAS; (e) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed.
- For a recent review, see: (a) M.-Y. Han, J.-Y. Jia and W. Wang, Tetrahedron Lett., 2014, 55, 784 CrossRef CAS PubMed . For recent examples, see: ; (b) X. Dou, F. Zhong and Y. Lu, Chem. – Eur. J., 2012, 18, 13945 CrossRef CAS PubMed; (c) K. E. Ozboya and T. Rovis, Chem. Sci., 2011, 2, 1835 RSC; (d) D. Enders, C. Wang and J. W. Bats, Angew. Chem., Int. Ed., 2008, 47, 7539 CrossRef CAS PubMed; (e) H. Li, L. Zu, H. Xie, J. Wang and W. Wang, Chem. Commun., 2008, 5636 RSC.
- For a recent review, see: (a) S. Goudedranche, W. Raimondi, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Synthesis, 2013, 1909 CAS . For recent examples, see: ; (b) Z. Jin, R. Yang, Y. Du, B. Tiwari, R. Ganguly and Y. R. Chi, Org. Lett., 2012, 14, 3226 CrossRef CAS PubMed; (c) L. Huo, A. Ma, Y. Zhang and D. Ma, Adv. Synth. Catal., 2012, 354, 991 CrossRef CAS; (d) H. Li, J. Zhao, L. Zeng and W. Hu, J. Org. Chem., 2011, 76, 8064 CrossRef CAS PubMed; (e) B. Wanner, J. Mahatthananchai and J. F. Bode, Org. Lett., 2011, 13, 5378 CrossRef CAS PubMed; (f) J. Jiang, J. Yu, X.-X. Sun, Q.-Q. Rao and L.-Z. Gong, Angew. Chem., Int. Ed., 2008, 47, 2458 CrossRef CAS PubMed; (g) N. Li, X.-H. Chen, J. Song, S.-W. Luo, W. Fan and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 15309 Search PubMed.
- For recent domino synthesis of seven-membered heterocycles in the racemic series, see: (a) J. Zhou and Y.-Y. Yeung, Org. Lett., 2014, 16, 2134 CrossRef CAS PubMed; (b) J.-M. Yang, C.-Z. Zhu, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 5142 CAS; (c) I. Nakamura, Y. Kudo and M. Terada, Angew. Chem., Int. Ed., 2013, 52, 7536 CrossRef CAS PubMed; (d) Y. A. Cheng, T. Chen, C. K. Tan, J. J. Heng and Y.-Y. Yeung, J. Am. Chem. Soc., 2012, 134, 16492 CrossRef CAS PubMed; (e) S. Li and J. Wu, Chem. Commun., 2012, 48, 8973 RSC.
- For reviews on general approaches to synthesize seven-membered heterocycles, see: (a) P. A. Evans and B. Holmes, Tetrahedron, 1991, 47, 9131 CrossRef CAS; (b) G. Rousseau and F. Homsi, Chem. Soc. Rev., 1997, 26, 453 RSC.
- G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95 CrossRef CAS.
- (a) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed; (b) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed.
- Y. Wang, M.-S. Tu, F. Shi and S.-J. Tu, Adv. Synth. Catal., 2014, 356, 2009 CrossRef CAS.
- (a) A. Michaut, S. Miranda-García, J. C. Menéndez, Y. Coquerel and J. Rodriguez, Eur. J. Org. Chem., 2008, 4988 CrossRef CAS; (b) F. López and J. L. Mascareñas, Chem. – Eur. J., 2007, 13, 2172 CrossRef PubMed; (c) H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef CAS PubMed.
- For the use of α-ketoamides in organocatalyzed transformations, see: (a) M. M. Sanchez Duque, S. Goudedranche, A. Quintard, T. Constantieux, X. Bugaut, D. Bonne and J. Rodriguez, Synthesis, 2013, 1659 Search PubMed; (b) C. Joie, K. Deckers and D. Enders, Synthesis, 2014, 799 Search PubMed; (c) C. Joie, K. Deckers, G. Raabe and D. Enders, Synthesis, 2014, 1539 Search PubMed.
- For examples of organocatalyzed Michael–hemiaminalization reactions, see: (a) R. Rios, I. Ibrahem, J. Vesely, H. Sundén and A. Córdova, Tetrahedron Lett., 2007, 48, 8695 CrossRef CAS PubMed; (b) W. Zhanga and J. Franzén, Adv. Synth. Catal., 2010, 352, 499 CrossRef; (c) G. Valero, J. Schimer, I. Cisarova, J. Vesely, A. Moyano and R. Rios, Tetrahedron Lett., 2009, 50, 1943 CrossRef CAS PubMed; (d) X. Wu, Q. Liu, H. Fang, J. Chen, W. Cao and G. Zhao, Chem. – Eur. J., 2012, 18, 12196 CrossRef CAS PubMed.
- D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Chem. – Eur. J., 2013, 19, 2218 CrossRef CAS PubMed.
- (a) C. Schöpf and K. Koch, Justus Liebigs Ann. Chem., 1942, 552, 37 CrossRef; (b) G. Habermehl, Chem. Ber., 1966, 99, 1439 CrossRef CAS; (c) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS PubMed; (d) For a recent review on other dihereobicyclo[3.2.1]octanes, see: M. F. Flores and D. Díez, Synlett, 2014, 1643 Search PubMed.
- (a) A. Padwa, L. Precedo and M. A. Semones, J. Org. Chem., 1999, 64, 4079 CrossRef CAS; (b) L. Ollero, L. Castedo and D. Dominguez, Tetrahedron, 1999, 55, 4445 CrossRef CAS.
- X.-D. Wu, L. Wang, J. He, X.-Y. Li, L.-B. Dong, X. Gong, X. Gao, L.-D. Song, Y. Li, L.-Y. Peng and Q.-S. Zhao, Helv. Chim. Acta, 2013, 96, 2207 CrossRef CAS.
- (a) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS PubMed; (b) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821 RSC.
- Below this temperature of −7 °C, the reaction medium tends to freeze.
- For complete reaction optimization, see the ESI†.
- As already observed in previous studies from our group, N-alkyl substituted 1,2-ketoamides are unproductive in this transformation, see: O. Baslé, W. Raimondi, M. M. Sanchez Duque, D. Bonne, T. Constantieux and J. Rodriguez, Org. Lett., 2010, 12, 5246 CrossRef PubMed.
- CCDC 1015393.
- Y. Xu, S. Matsunaga and M. Shibasaki, Org. Lett., 2010, 12, 3246 CrossRef CAS PubMed.
- P. Kulanthaivel, Y. F. Hallock, C. Boros, S. M. Hamilton, W. P. Janzen, L. M. Ballas, C. R. Loomis, J. B. Jiang and B. Katz, J. Am. Chem. Soc., 1993, 115, 6452 CrossRef CAS.
- (a) Y.-F. Song, Y. Qu, X.-P. Cao and W. Zhang, Mar. Biotechnol., 2011, 13, 868 CrossRef CAS PubMed; (b) E. Torres-Marquez, J. Sinnett-Smith, S. Guha, R. Kui, R. T. Waldron, O. Rey and E. Rozengurt, Biochem. Biophys. Res. Commun., 2010, 391, 63 CrossRef CAS PubMed; (c) J. Hu and M. J. Miller, Tetrahedron Lett., 1995, 36, 6379 CrossRef CAS.
- D. M. Floyd, S. D. Kimball, J. Krapcho, J. Das, C. F. Turk, R. V. Moquin, M. W. Lago, K. J. Duff, V. G. Lee, R. E. White, R. E. Ridgewell, S. Moreland, R. J. Brittain, D. E. Normandin, S. A. Hedberg and G. G. Cucinotta, J. Med. Chem., 1992, 35, 756 CrossRef CAS.
- (a) J. C. Martin and R. J. Arhart, J. Am. Chem. Soc., 1971, 93, 4327 CrossRef CAS; (b) R. J. Arhart and J. C. Martin, J. Am. Chem. Soc., 1972, 94, 5003 CrossRef CAS; (c) J. C. Martin, R. J. Arhart, R. Franz, E. Perozzi and L. Kaplan, Org. Synth., 1997, 57, 22 Search PubMed.
- A. C. Breman, J. Dijkink, J. H. van Maarseveen, S. S. Kinderman and H. Hiemstra, J. Org. Chem., 2009, 74, 6327 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and spectral data for new compounds as well as optimization of the reaction conditions. CCDC 1015393. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07731h |
| This journal is © The Royal Society of Chemistry 2014 |