
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed enantioselective 1,4-addition of alkyl groups to N-sulfonyl imines†
Johannes
Westmeier
and
Paultheo
von Zezschwitz
*
Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35032 Marburg, Germany. E-mail: zezschwitz@chemie.uni-marburg.de
First published on 25th September 2014
Abstract
In copper(I)/phosphoramidite-catalyzed asymmetric 1,4-additions of dialkylzinc, N-sulfonyl imines are more reactive and furnish higher enantiomeric excesses than the respective cycloalk-2-enones. This enables formation of a quaternary stereocenter as well as a cis-selective addition to an imine derived from 5-methylcyclohex-2-enone. The 1,4-adducts can be transformed in stereodivergent reductions yielding cis- or trans-3-alkylcycloalkyl amides.
The copper-catalyzed enantioselective 1,4-addition of organometallic reagents to α,β-unsaturated acceptors is a fundamental transformation in organic synthesis.1 Originally reported with dialkylzinc, it has been developed towards the use of other types of nucleophiles such as organoaluminium reagents,2 Grignard reagents,3 and zirconocenes4 as well as towards addition of unsaturated residues such as aryl and alkenyl groups.5 While the latter can also be efficiently performed under rhodium catalysis,6 use of copper is highly attractive due to its lower price. Moreover, much effort is being spend on the study of 1,4-additions to β,β-disubstituted compounds to create quaternary stereocenters.7 In the case of unactivated, plain enones, this can be achieved by the use of more reactive aluminium or Grignard reagents and/or more reactive catalysts with NHC ligands.8
In contrast to carbonyl compounds, α,β-unsaturated imines have hardly been used for such reactions9,10 which is surprising in view of the enormous importance of nitrogen-containing moieties in natural and artificial bioactive molecules. Tomioka et al. reported on the 1,4-addition of dialkylzinc to N-sulfonyl imines derived from cinnamaldehyde and achieved up to 91% ee using a Cu/amidophosphane catalyst if the sulfonyl group carried a bulky aryl substituent.9a Carretero et al. studied the 1,4-addition of ZnMe2 to N-sulfonyl imines derived from chalcones. Up to 80% ee was achieved using a Cu/binol-phosphoramidite catalyst in the case of N-2-pyridylsulfonyl imines, while no conversion occurred with the respective tosyl derivatives.9b Finally, Palacios et al. reported up to 88% ee in the 1,4-addition of ZnEt2 to acyclic β,γ-unsaturated N-aryl α-iminoesters using a Cu/taddol-phosphoramidite catalyst.9c In all reports, the 1,4-adducts were either hydrolyzed to the respective carbonyls or transformed by oxidative cleavage of the C,C-double bonds in the tautomeric enamines. Subsequent transformation to amines was reported in only one example in which hydrogenation over Pd/C furnished an 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 mixture of diastereomers.9c
:18 mixture of diastereomers.9c
Recently, we reported the first preparation of cycloalk-2-enone-derived N-sulfonyl imines11 as well as their transformation in highly enantioselective Rh(I)/binap-catalyzed additions of methyl- and arylaluminium reagents. The 1,2- versus 1,4-selectivity of this reaction was influenced by several factors, and we initially performed optimization towards 1,2-addition to deliver valuable α-tertiary cycloalk-2-enyl amides.12 Moreover, we developed regio- and enantioselective Rh(I)/binap-catalyzed 1,4-additions of arylzinc halides to these substrates. After subsequent stereodivergent reduction, cis- or trans-3-arylcycloalkyl amides were obtained which can oxidatively be degraded to deliver 3-aminocycloalkanecarboxylic acids.13 Both 3-aryl- as well as 3-alkyl-substituted cycloalkyl amines are commonly found in pharmaceutically active molecules.14,15 Based on our previous results, a Rh-catalyzed enantioselective 1,4-addition of AlMe3 should be feasible, but we decided to study economically more attractive Cu-based catalysts. This communication describes the first highly regio- and enantioselective 1,4-additions of alkyl groups to cyclic N-sulfonyl ketimines with subsequent stereodivergent reduction. Catalyzed by a Cu–phosphoramidite complex, these transformations surpass those of the corresponding carbonyl derivatives in both reactivity and enantioselectivity.
As a model reaction, the addition of ZnEt2 to the cyclohex-2-enone-derived N-tosyl imine 1a was studied by applying classical conditions with Feringa's phosphoramidite L1 (Table 1).16 Using toluene as the solvent, excellent results were achieved from the very beginning, and a reaction temperature of −30 °C proved to be optimal, partial catalyst decomposition being observed at 0 °C and rt (entries 1–4). Moreover, some other solvents were screened but toluene appeared to be the most suitable solvent (entries 5–7 vs. 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | “Cu” | Solvent | T (°C) | t (h) | Yielda (%) | eeb (%) |
| a Determined by 1H NMR analysis of the crude product against diphenylmethane as an internal standard. b Determined by GC after hydrolysis to the respective ketone. c Without ligand L1. d Without the Cu salt and ligand L1. | ||||||
| 1 | Cu(OTf)2 | Toluene | −30 | 1 | 97 | 96 |
| 2 | Cu(OTf)2 | Toluene | −78 | 16 | 92 | 47 |
| 3 | Cu(OTf)2 | Toluene | 0 | 1 | 96 | 96 |
| 4 | Cu(OTf)2 | Toluene | rt | 1 | 92 | 92 |
| 5 | Cu(OTf)2 | CH2Cl2 | −30 | 1 | 98 | 91 |
| 6 | Cu(OTf)2 | Et2O | −30 | 1 | 87 | 96 |
| 7 | Cu(OTf)2 | THF | −30 | 1 | 86 | 51 |
| 8 | Cu(OTf)2c |
Toluene | −30 | 1 | 62 | 0 |
| 9 | —d | Toluene | −30 | 1 | 14 | 0 |
| 10 | Cu(OAc)2·2H2O | Toluene | −30 | 1 | 91 | 97 |
| 11 | CuCl | Toluene | −30 | 1 | 96 | 97 |
| 12 | (CuI)4(DMS)3 | Toluene | −30 | 1 | 88 | 97 |
| 13 | Cu(MeCN)4BF4 | Toluene | −30 | 1 | 78 | 97 |
| 14 | CuTC | Toluene | −30 | 1 | 91 | 97 |
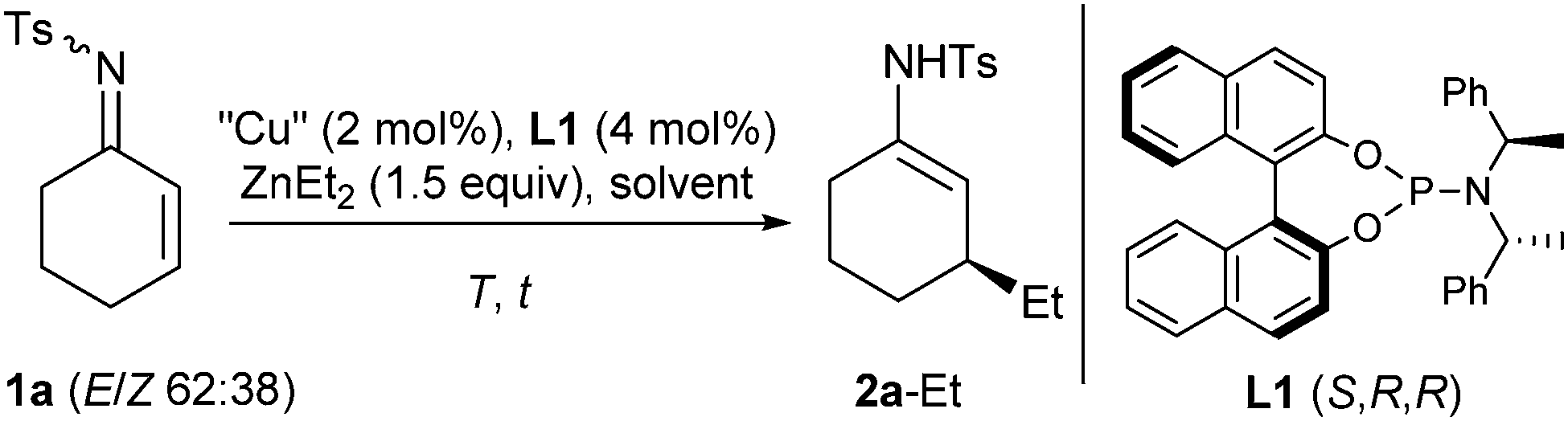
In the absence of the chiral ligand and even without the copper salt, some background reactivity was observed (entries 8 and 9), and screening of several copper(I) and copper(II) sources led to only slight variations in the yields (entries 10–14). In the end, CuTC (TC = thiophene-2-carboxylate) was chosen due to its chemical stability, and the reaction was monitored using continuous IR detection which revealed an induction period of about 0.5 min after addition of ZnEt2 to the catalyst–substrate mixture (see ESI†). In all these transformations, the 1,4-adduct was obtained exclusively as enamide 2a-Et, and not as the tautomeric imine, and the (E)- and the (Z)-isomer of substrate 1a underwent the reaction. The facial selectivity of this 1,4-addition is identical with both types of substrates, imine 1a and the respective enone, as proven by hydrolysis of 2a-Et and comparison with an authentic sample of (S)-3-ethylcyclohexanone.
Similar to the respective 3-aryl derivatives, enamide 2a-Et partially hydrolyzed upon attempted column chromatography, but could be transformed in stereodivergent reductions.13 While pure trans-3-ethylcyclohexyl amide 4a was obtained after transfer hydrogenation catalyzed by racemic RuCl(p-cymene)[Ts-DPEN]17 (rac-3), reduction with tBuNH2·BH3 furnished a 90:10 cis/trans mixture which could be separated by chromatography to deliver cis-4a-Et in a 64% yield (Table 2, entries 1 and 2). The catalyst loading could be reduced to 0.01 mol% (TON 8900) revealing the outstanding reactivity of N-tosyl imines in this transformation (entry 3).18 Moreover, good results were also achieved with the simplified ligand L24b (entry 4), and methyl addition proceeded equally well despite the notorious lower reactivity of ZnMe2 in comparison with that of ZnEt2 (entry 5). In contrast, aluminium reagents appeared to be less suitable for this transformation (entries 6 and 7).2 Besides N-tosyl imine 1a, the N-tert-butylsulfonyl imine 5a could also be reacted, yet the reactivity and the chemoselectivity were inferior (entry 8). Nevertheless, these types of substrates are synthetically useful, because the tert-butylsulfonyl group can be cleaved under acidic conditions.19 The N-phosphinoyl imine 7a, however, furnished a racemic 1,4-adduct (entry 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | “RM” | Product | t (h) | dra (trans/cis) | Yieldb (%) | eec (%) |
| a Determined by 1H NMR analysis of the crude product. b Isolated yield of the diastereomerically pure product. c Determined by HPLC. d Reduction performed with tBuNH2·BH3 in CH2Cl2. e Performed with 0.01 mol% CuTC and 0.02 mol% L1. f L2 as the ligand. g 1,4-Adddition in Et2O. h Reduction performed with L-selectride in THF. | ||||||
| 1 | ZnEt2 | 4a-Et | 1 | >97:3 |
85 | 98 |
| 2 | ZnEt2 | 4a-Et | 1 | 10:90d |
64 | 96 |
| 3e | ZnEt2 | 4a-Et | 1 | >97:3 |
89 | 87 |
| 4f | ZnEt2 | 4a-Et | 1 | >97:3 |
83 | 91 |
| 5 | ZnMe2 | 4a-Me | 1 | >97:3 |
87 | 98 |
| 6g | AlEt3 | 4a-Et | 1.5 | — | 0 | — |
| 7g | AlMe3 | 4a-Me | 1.5 | >97:3 |
28 | 96 |
| 8 | ZnEt2 | 6a-Et | 20 | >97:3 |
60 | 95 |
| 9 | ZnEt2 | 8a-Eth | 20 | >97:3 |
62 | 0 |

Besides the cyclohex-2-enone-derived substrates, various additional imines were transformed in this addition-reduction sequence (Table 3). Starting with the N-tert-butylsulfonyl imine 5b20 derived from cyclopentenone, the respective cycloalkyl amide 6b-Et was obtained as a 23:77 trans/cis mixture after transfer hydrogenation with the racemic Ru-catalyst 3 (entry 1). The use of the (S,S)-enantiomer of 3 led to a significantly higher yield and also to a slight increase of enantiopurity (entry 2). At first, this 64–69% ee appeared to be insufficient, however, given that ligand L1 leads to only 10% ee with cyclopent-2-enone,16 these results reveal a significantly higher stereoinduction in the case of N-sulfonyl imines. Transformation of the cycloheptenone-derived imine 5c20 again proceeded with very high enantio- and diastereoselectivity (entry 3). N-Tosyl imines 1b and 1c with a geminal disubstitution vicinal to the C,C-double bond underwent the 1,4-addition with a similar efficiency as the related enones,16,21 the 1,4-adduct 2b-Et was reduced with NaBH4 to furnish the cis-cyclopentyl amide 4b-Et (entries 4 and 5). Good to very good enantioselectivities were also achieved in the case of substrates 1d and 1e with a geminal disubstitution vicinal to the C,N-double bond, and the 1,4-adducts were reduced with NaBH4 or tBuNH2·BH3 due to failure of the transfer hydrogenation (entries 6 and 7). Transformation of the 5,5-dimethyl substituted compound 1f again demonstrated a higher enantioselectivity compared to that of the respective enone (entry 8).16
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Imine | t (h) | Reductiona | drb (trans/cis) | Yieldc (%) | eed (%) |
| a A: racemic RuCl(p-cymene)[Ts-DPEN] (rac-3), HCOOH/NEt3 (5/2), CH3CN; B: As in A, but (S,S)-3; C: NaBH4, EtOH; D: tBuNH2·BH3, CH2Cl2. b Determined by 1H NMR analysis of the crude product. c Isolated yield. d Determined by HPLC; values in parentheses were reported for transformation of the respective enone using the same chiral ligand. e Performed at −15 °C with 10 mol% CuTC and 12 mol% ent-L1. f Isolated as enamide 2h-Et. | ||||||
| 1 | 5b | 24 | A | 23:77 |
52 | 64 (10) |
| 2 | 5b | 20 | B | 23:77 |
72 | 69 |
| 3 | 5c | 16 | A | 96:4 |
66 | ∼92 (>98) |
| 4 | 1b | 5 | C | 7:93 |
70 | 91 |
| 5 | 1c | 18 | A | >97:3 |
87 | 96 (>98) |
| 6 | 1d | 20 | C | 64:36 |
89 | 80 |
| 7 | 1e | 16 | D | <3:97 |
76 | 91 |
| 8 | 1f | 1 | A | >97:3 |
83 | 94 (84–88) |
| 9 | 1g | 20e | B | 82:18 |
54 | 94 |
| 10 | 1h | 2.5 | —f | — | 68 | 72 (75) |

With an increased catalyst loading and at increased temperature, 82% conversion was achieved in the 1,4-addition to the 3-methyl substituted imine 1g, and hydrolysis of the 1,4-adduct delivered the respective ketone with 86% ee. Towards formation of the cycloalkyl amide trans-4g-Et, best results were obtained when performing the 1,4-addition with the (R,S,S)-enantiomer of ligand L1 and the transfer hydrogenation with (S,S)-3. This combination is obviously the matched pair and led to a slight amplification of the enantiopurity to 94% ee and to a good 82:18 dr (entry 9). Thus, this protocol even enables construction of quaternary stereocenters which is highly remarkable because copper–phosphoramidite complexes fail to catalyze ZnEt2 addition to unactivated β,β-disubstituted enones, more reactive organometallic reagents and/or catalysts being needed.7,8 As an example for an acyclic substrate, the chalcone-derived N-tosyl imine 1h was converted, and its 1,4-adduct proved to be more stable than those of the cyclic imines. Thus, the enamide 2h-Et could be purified by column chromatography and was obtained in a good yield with 72% ee (entry 10). This compares well with the 75% ee reported by Feringa et al. for transformation of the chalcone itself at −25 °C22 and the 60% ee reported by Carretero et al. for transformation of the N-2-pyridylsulfonyl imine of the chalcone at –78 °C,9b both with the same ligand L1.
Finally, remarkable results were obtained with imine 1i which was prepared from (R)-5-methylcyclohex-2-enone (Scheme 1): copper-catalyzed 1,4-additions to such 5-alkyl-substitued cyclohex-2-enones are always dominated by a very strong trans-directing substrate control, which cannot be overcome by catalyst control.23 Thus, it is not possible to obtain cis-3,5-dialkyl substituted cyclohexanones from Cu-catalyzed 1,4-additions. This substrate control was also observed in the case of imine (R)-1i: using a racemic phosphoramidite ligand in the 1,4-addition, the enamide trans-2i-Et was formed as a single diastereomer. With ligand L1, however, a 48:52 mixture of the trans- and cis-configured 1,4-adducts was obtained, reflecting comparable strength of the substrate and catalyst control. Transfer hydrogenation of this diastereomeric mixture delivered exclusively (1S,3S,5R)-4i-Et from the cis-configured enamide, while a 54:46 mixture of epimers was obtained from the trans-diastereomer.
 | ||
| Scheme 1 Synthesis of the cis-3,5-disubstituted amide (1S,3S,5R)-4i-Et. | ||
tert-Butylsulfonyl groups can readily be removed under acidic conditions,19 while cleavage of tosyl groups frequently requires rather harsh conditions. After Boc protection, however, the detosylation of amide trans-4a-Et occurred under mild conditions in a high yield (Scheme 2),24 which proves the general synthetic applicability of this 1,4-addition-reduction sequence.
 | ||
| Scheme 2 Detosylation of cyclohexyl amide 4a. | ||
In summary, the 1,4-addition of dialkylzinc reagents to cyclic N-sulfonyl imines outpaces transformations of the respective enones in both reactivity and stereoselectivity. This is highlighted by the formation of a cis-3,5-dialkyl substituted adduct from imine 1i and the formation of a quaternary stereocenter from imine 1g. While some imines furnished less than 90% ee with phosphoramidite L1, excellent enantioselectivities have been reported for transformations of the respective enones applying other chiral ligands.25 Thus, better selectivities can certainly be achieved with these imines, too. Anyway, the broad scope of applicable cyclic imines and the stereodivergence of the subsequent reduction offer a highly flexible access to synthetically and biologically important 3-alkylcycloalkyl amines. We are now working on employing other organometallic reagents in this copper-catalyzed 1,4-addition and on using the initially formed zinc aza-enolates for subsequent C,C-bond formations.
The authors are indebted to Jan Herritsch and Christoph Priem, Philipps-Universität Marburg, for technical assistance and to the BASF SE, Ludwigshafen for the generous donation of chemicals. J. W. thanks the Konrad-Adenauer-Stiftung, Sankt Augustin, for a scholarship.
Notes and references
- (a) A. Alexakis and C. Benhaim, Eur. J. Org. Chem., 2002, 3221 CrossRef CAS; (b) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796 CrossRef CAS PubMed; (c) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039 RSC; (d) Copper-Catalyzed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 2014 Search PubMed.
- A. Alexakis, V. Albrow, K. Biswas, M. d'Augustin, O. Prieto and S. Woodward, Chem. Commun., 2005, 2843 RSC.
- (a) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed; (b) T. Robert, J. Velder and H.-G. Schmalz, Angew. Chem., Int. Ed., 2008, 47, 7718 CrossRef CAS PubMed.
- (a) R. M. Maksymowicz, P. M. C. Roth and S. P. Flechter, Nat. Chem., 2012, 4, 649 CrossRef CAS PubMed; (b) M. Sidera, P. M. C. Roth, R. M. Maksymowicz and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 7995 CrossRef CAS PubMed.
- (a) D. Pena, F. Lopez, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2004, 1836 RSC; (b) D. Müller and A. Alexakis, Chem. Commun., 2012, 48, 12037 RSC.
- (a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed; (b) K. Yoshida and T. Hayashi, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005, p. 55 Search PubMed; (c) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95 CrossRef CAS.
- C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC.
- (a) M. d'Augustin, L. Palais and A. Alexakis, Angew. Chem., Int. Ed., 2005, 44, 1376 CrossRef PubMed; (b) K.-s. Lee, M. K. Brown, A. W. Hird and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 7182 CrossRef CAS PubMed; (c) D. Martin, S. Kehrli, M. d'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416 CrossRef CAS PubMed; (d) T. L. May, M. K. Brown and A. H. Hoveyda, Angew. Chem., Int. Ed., 2008, 47, 7358 CrossRef CAS PubMed; (e) C. Hawner, K. Li, V. Cirriez and A. Alexakis, Angew. Chem., Int. Ed., 2008, 47, 8211 CrossRef CAS PubMed.
- (a) T. Soeta, M. Kuriyama and K. Tomioka, J. Org. Chem., 2005, 70, 297 CrossRef CAS PubMed; (b) J. Esquivias, R. G. Arrayás and J. C. Carretero, J. Org. Chem., 2005, 70, 7451 CrossRef CAS PubMed; (c) F. Palacios and J. Vicario, Org. Lett., 2006, 8, 5405 CrossRef CAS PubMed.
- For chiral auxiliary-based diastereoselective 1,4-additions of cuprates to imines, see: (a) J. P. McMahon and J. A. Ellman, Org. Lett., 2005, 7, 5393 CrossRef CAS PubMed; (b) K. Sammet, C. Gastl, A. Baro, S. Laschat, P. Fischer and I. Fettig, Adv. Synth. Catal., 2010, 352, 2281 CrossRef CAS.
- S. Hirner, J. Westmeier, S. Gebhardt, C. H. Müller and P. von Zezschwitz, Synlett, 2014, 1697 Search PubMed.
- S. Hirner, A. Kolb, J. Westmeier, S. Gebhardt, S. Middel, K. Harms and P. von Zezschwitz, Org. Lett., 2014, 16, 3162 CrossRef CAS PubMed.
- S. Gebhardt, C. H. Müller, J. Westmeier, K. Harms and P. von Zezschwitz, Adv. Synth. Catal. Search PubMed , submitted.
- A reaxys survey revealed more than 400 patents comprising 3-alkylcyclohexyl amines.
- 3-Alkyl-substituted cyclohexyl amines can be prepared by an organocatalytic reaction cascade. For enantioselective formation of the cis-diastereomers, see (a) J. Zhou and B. List, J. Am. Chem. Soc., 2007, 129, 7498 CrossRef CAS PubMed . For racemic preparation of the trans-diastereomers, see: ; (b) J. Zhou and B. List, Synlett, 2007, 2037 Search PubMed.
- B. L. Feringa, Acc. Chem. Res., 2000, 33, 346 CrossRef CAS PubMed.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- A TON >3000 was mentioned by Feringa for transformations of cyclic enones, see ref. 16.
- P. Sun, S. M. Weinreb and M. Shang, J. Org. Chem., 1997, 62, 8604 CrossRef CAS.
- The respective N-tosyl imine is problematic to prepare, see ref. 11.
- For the transformation of 4,4-dialkoxy-substituted cyclopentenones, see: L. A. Arnold, R. Naasz, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2002, 67, 7244 CrossRef CAS PubMed.
- L. A. Arnold, R. Imbos, A. Mandoli, A. H. M. de Vries, R. Naasz and B. L. Feringa, Tetrahedron, 2000, 56, 2865 CrossRef CAS.
- (a) R. Naasz, L. A. Arnold, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 927 CrossRef CAS; (b) T. Soeta, K. Selim, M. Kuriyama and K. Tomioka, Tetrahedron, 2007, 63, 6573 CrossRef CAS PubMed.
- General procedure: B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017 RSC.
- For highly enantioselective ZnEt2 additions to the enones corresponding to imines 1d and 5b, see: (a) S. J. Degrado, H. Mizutani and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 755 CrossRef CAS ; for the enone corresponding to 1h, see: ; (b) X. Hu, H. Chen and X. Zhang, Angew. Chem., Int. Ed., 1999, 38, 3518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, and NMR spectra for all new compounds. See DOI: 10.1039/c4cc07134d |
| This journal is © The Royal Society of Chemistry 2014 |