
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transformation from non- to double-interpenetration in robust Cd(II) doubly-pillared-layered metal–organic frameworks†
Himanshu
Aggarwal
,
Prem
Lama
and
Leonard J.
Barbour
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Private Bag X1, Matieland, 7602, South Africa. E-mail: ljb@sun.ac.za
First published on 7th October 2014
Abstract
Two known Cd(II) non-interpenetrated doubly-pillared metal organic frameworks are shown to undergo a change of degree of interpenetration upon loss of lattice solvent molecules to yield doubly-interpenetrated frameworks. The conversion is inferred from powder X-ray diffraction (PXRD) patterns and supported by additional evidence from single-crystal diffraction (SCD) data.
We1 and others2 have long been interested in solid–solid phase transformations resulting from guest insertion/removal into/from inclusion compounds. Indeed, such phenomena are particularly important for the utility of porous crystalline systems such as metal–organic frameworks (MOFs). Some of the most important attributes of a MOF include its rigidity, free volume and surface area. Typically, a MOF would be prepared3 (usually under solvothermal conditions) and then ‘activated’ by removal of the solvent molecules. In ideal cases the material would retain its framework connectivity without collapse or distortion of the network – MOFs with metal cluster nodes (i.e. secondary building units) are generally thought to be more robust to the activation process.4
Interpenetration5 or catenation of MOFs6 is a well-known phenomenon; when the geometry of the system allows, two or more independent frameworks can become interwoven to yield double-, triple- or even higher-order interpenetration. For a given framework the free volume and surface area depend on the total number of nets involved. Since gas sorption7 is considered one of the major applications of MOFs, interpenetration is usually undesirable because it results in reduction of the guest-accessible space8 (i.e. less porosity). To overcome this problem, several groups have tried to control the degree of interpenetration using various techniques such as liquid epitaxy,9 variation of temperature10,11 and concentration,11 and the use of bulkier solvents for crystallisation.12 As a result of these strategies, a number of MOFs have been reported that exhibit less interpenetration than their network topologies can allow.13 However, the phenomenon of dynamic conversion between different orders of interpenetration has received little attention to date.
We recently reported how a highly porous doubly-interpenetrated MOF converts to a less porous triply-interpenetrated form upon loss of solvent molecules from the channels.14 Since it seems that switching of degree of interpenetration upon activation is generally assumed to be improbable, we became interested in investigating whether such occurrences are restricted to only a handful of specific systems, or if the phenomenon might be more general. We thus expanded our study to include a supposedly more robust system, and its analogue.
Here we report on our investigation of two closely-related non-interpenetrated MOFs which have previously been described by other groups. Frameworks 1 and 2 have the formulae [Cd(tp)(4,4′-bpy)]11,15 and [Cd(atp)(4,4′-bpy)],10 respectively (Scheme 1, tp = terephthalate; 4,4′-bpy = 4,4′-bipyridine and atp = 2-aminoterephthalate).
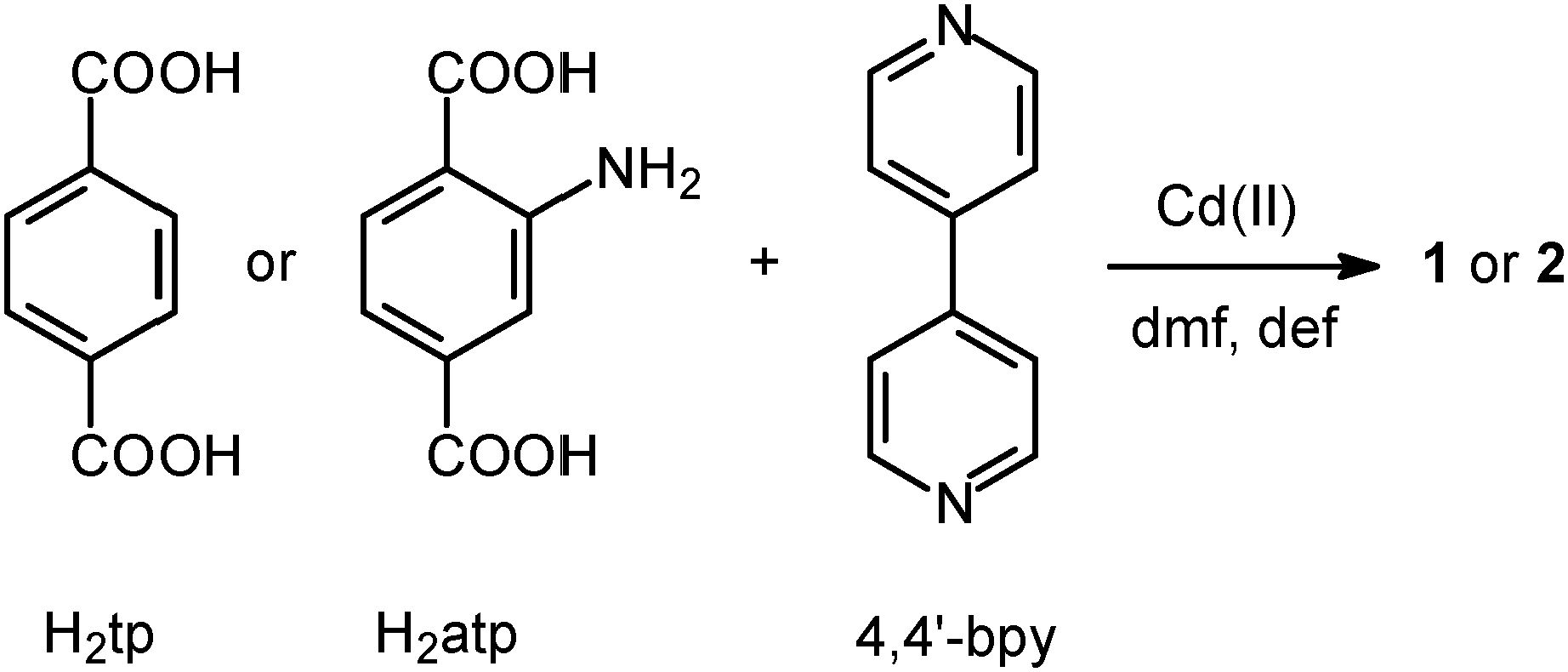 | ||
| Scheme 1 Synthetic scheme for the preparation of frameworks 1 and 2. | ||
These frameworks are comprised of Cd(II) ions coordinated to carboxylate linkers to form two dimensional networks, which are further linked by means of 4,4′-bipyridine ligands to yield infinite three-dimensional doubly-pillared structures. Both systems have been reported to form non-interpenetrated as well as doubly-interpenetrated structures (Fig. 1). However, the possibility has not been suggested that the corresponding structures might interconvert by switching their degrees of interpenetration.
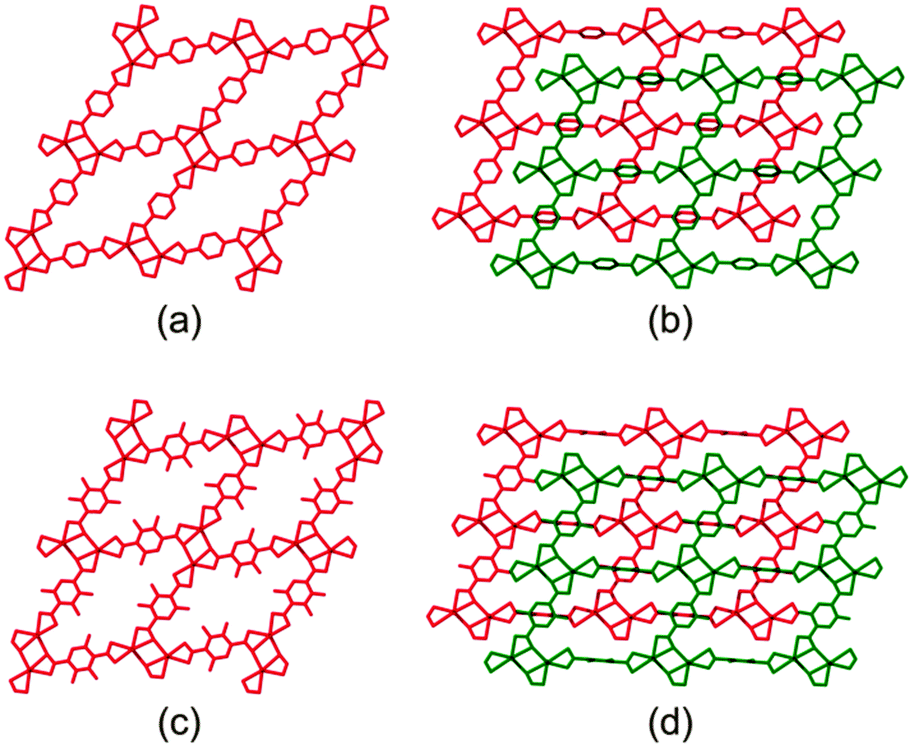 | ||
| Fig. 1 Packing diagrams of (a) non-interpenetrated 1, (b) doubly-interpenetrated 1, (c) non-interpenetrated 2 and (d) doubly-interpenetrated 2. | ||
As expected, the non-interpenetrated structures are more porous than their corresponding doubly-interpenetrated forms. As part of our investigation we prepared the non-interpenetrated form of framework 1 using the reported procedure.11 The crystal structure possesses free volume of 59.3% of the unit cell11,16 with dimethyl formamide (dmf) and water molecules occupying the channels. Thermogravimetric analysis (TGA) shows continuous weight loss up to 160 °C, which corresponds to the loss of all the solvent molecules. The PXRD pattern of the bulk material was compared with that simulated from the reported SCD structure11 to establish phase purity of the crystals. In order to completely remove the solvent molecules from the channels, the as-synthesised crystals were evacuated at 150 °C under dynamic vacuum for 12 hours. The crystals remained transparent upon desolvation, yielding a PXRD pattern different from that of the as-synthesised material but matching that simulated from the reported twofold-interpenetrated crystal structure of 115 (Fig. 2). This finding indicates that the as-synthesised non-interpenetrated framework transforms to its doubly-interpenetrated form upon activation.
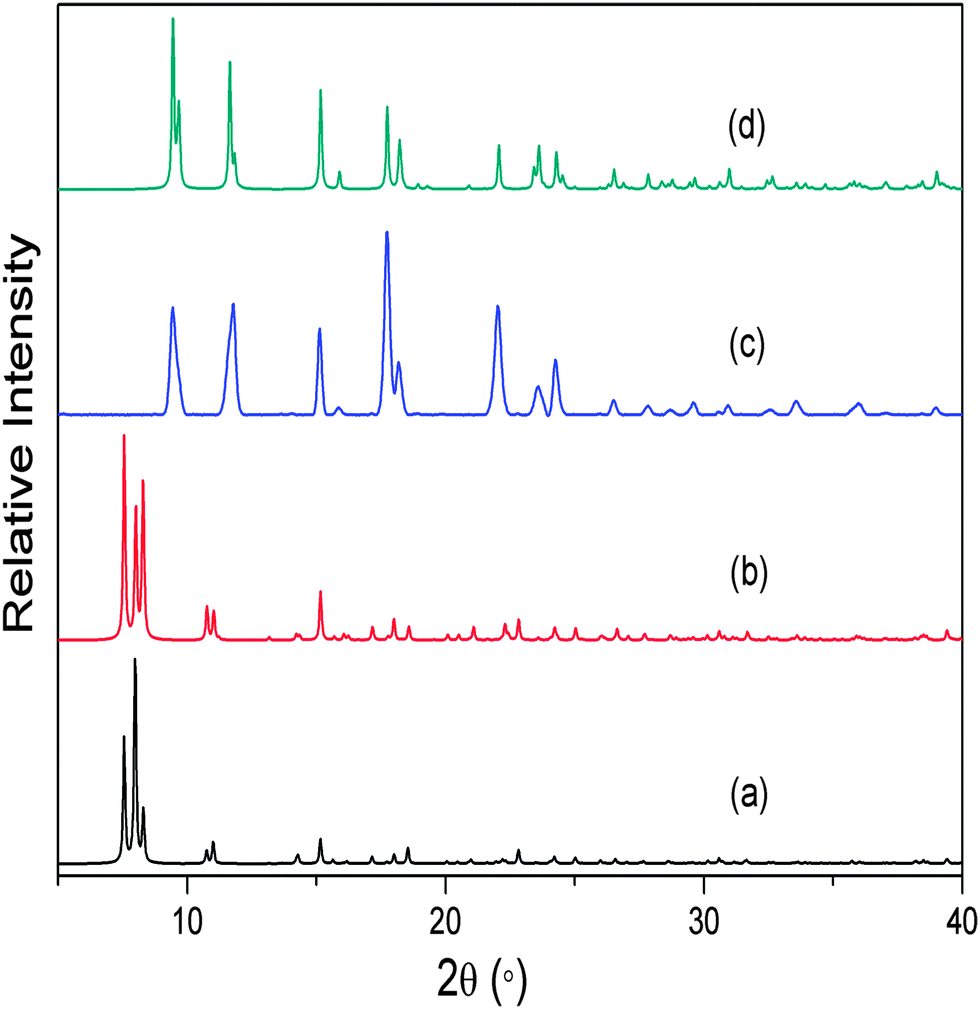 | ||
| Fig. 2 (a) Simulated PXRD pattern of non-interpenetrated 1, (b) experimental PXRD pattern of as-synthesised 1, (c) experimental PXRD pattern of 1 activated at 150 °C and (d) simulated PXRD pattern of the reported crystal structure of doubly-interpenetrated 1. | ||
This result is consistent with our previous observation that the loss of solvent from MOF channels can result in switching of degree of interpenetration, showing that the phenomenon is not limited to only one system. Although we attempted to record SCD intensity data for the activated crystals, they had been subjected to a relatively high activation temperature and the quality of the data was therefore insufficient for acceptable structure solution and refinement. Nevertheless, comparison of the unit cell parameters of the activated crystals with those of the reported twofold-interpenetrated structure confirmed that the crystals had indeed undergone a change in degree of interpenetration (ESI†). It seems reasonable to propose that the change in interpenetration occurs as a result of the empty space created upon solvent removal – the architecture of the framework can accommodate interpenetration and the coordination linkages are sufficiently labile to allow the process to proceed once the space becomes available.
As a continuation of this study, we also investigated the closely analogous system 2, where terephthalate is replaced by 2-aminoterephthalate, but the overall coordination environment around the central metal atoms remains the same. We prepared crystals of non-interpenetrated [Cd(atp)(4,4′-bpy)] by following the reported procedure;10 the PXRD pattern of the as-synthesised crystals was compared with the simulated PXRD pattern for the known structure in order to confirm phase purity of the bulk material. The free volume in framework 2 is 61.2% of the unit cell10,16 and TGA shows continuous weight loss until 220 °C, corresponding to the complete loss of dmf and water molecules from the channels. Similar to the treatment of 1, as-synthesised bulk crystals of 2 were evacuated at 150 °C under dynamic vacuum for 12 hours. The PXRD pattern of the resulting material differed considerably from that of the as-synthesised crystals (ESI†) but it did not match that simulated for the reported doubly-interpenetrated SCD structure of 2.10 Further activation was carried out at 270 °C, after which the PXRD pattern matched that of 2 (Fig. 3). The harsher conditions required for the activation of 2 can be attributed to the presence of the amino groups directed towards the framework channels; i.e. more energy is required for the process of solvent removal and concomitant disassembly/reassembly of the framework. As noted for the transformation of 1, it was possible to determine the unit cell parameters of 2 by means of SCD data, but the diffraction record was of insufficient quality for rigorous crystal structural analysis (ESI†).
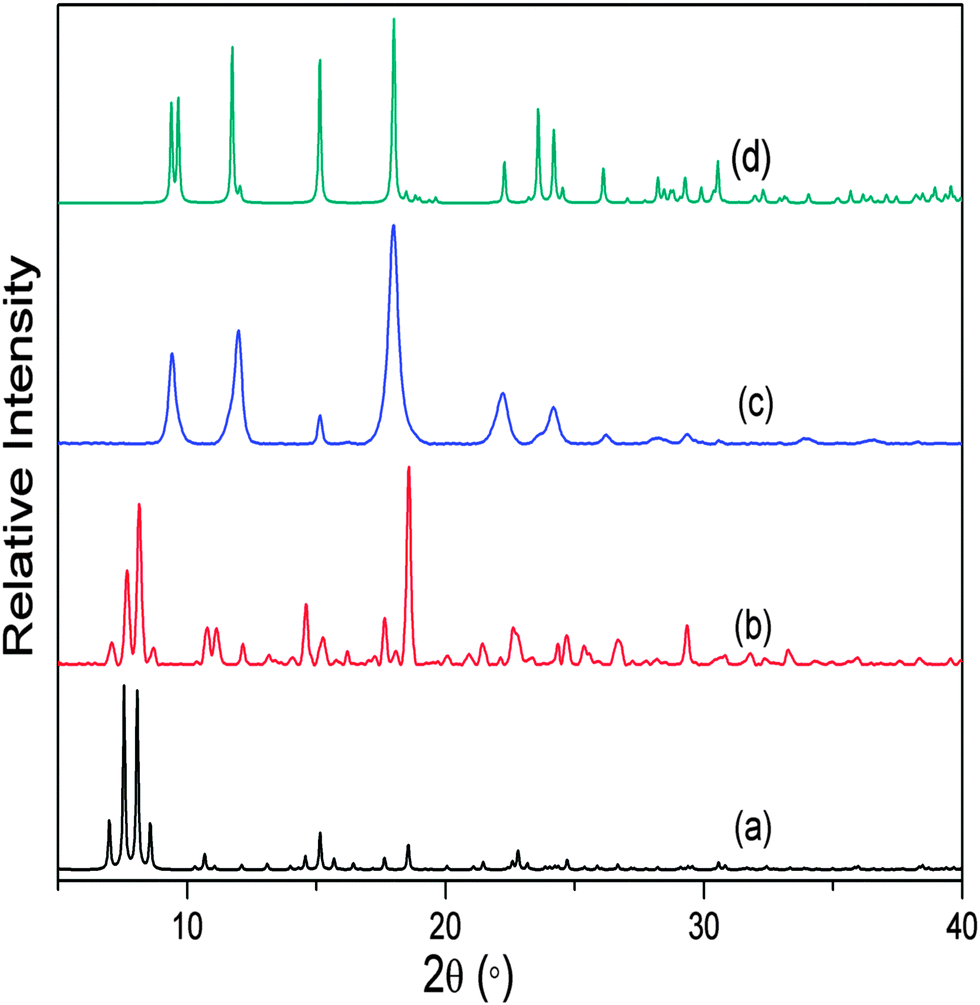 | ||
| Fig. 3 (a) Simulated PXRD pattern of non-interpenetrated 2, (b) experimental PXRD pattern of as-synthesised 2, (c) experimental PXRD pattern of 2 activated at 270 °C and (d) simulated PXRD pattern of the reported crystal structure of doubly-interpenetrated 2. | ||
Based on our previous report on the change in mode of interpenetration of [Zn2(ndc)2(4,4′-bpy)],14 we can postulate a possible mechanism for the transformations experienced by 1 and 2. It is well known that molecular crystal solvates are generally stabilised by the presence of the solvent molecules. That is, the solvent molecules play a structural role until they are removed, whereupon the host molecules rearrange to form a closely-packed apohost phase. We believe that the same holds true for some MOF structures; i.e. that the frameworks are partially supported by the solvent molecules. When an attempt is made to activate the crystals by removing the solvent molecules, the vacant spaces thus created allow the coordination bonds to become distorted. In extreme cases, the coordination environment about the metal centre can undergo modification.17 In even more severe cases, the secondary building units may disassemble and reassemble, which could also involve changes in entanglement of the coordination networks.14,18 Such a process involves rapid bond dissociation and formation to yield a thermodynamically more stable structure with a higher degree of interpenetration. It is interesting to note the differences between the current systems and our previously reported examples where the change of interpenetration was from doubly- to triply- in singly-pillared-layered paddlewheel structures. Systems 1 and 2 are more robust owing to their doubly-pillared-layered structures and the change in interpenetration is from non- to doubly-. Moreover, in the case of [Zn2(ndc)2(4,4′-bpy)]14 the conversion took place under ambient conditions whereas the transformation requires elevated temperatures for 1 and 2.
Close inspection of the non-interpenetrated [Cd(tp)(4,4′-bpy)] structure shows that four dinuclear Cd(II) clusters along with four tp units form a cyclic metallo-organic unit with cluster C1 in the same orientation as C3 and cluster C2 in the same orientation as C4 (Fig. 4). All of the aromatic units of the terephthalate ligands (L1 to L4) are in the metal–carboxylate plane – i.e. parallel to (001) and the doubly-pillaring bipyridyl linkers are parallel to [001], with a dihedral angle of 2.1(1)° between the two pyridyl rings. Upon activation of non-interpenetrated 1 at 150 °C, rotation of two of the four terephthalate linkers (L2 and L4) takes place. This is concomitant with coordination bond breaking and formation within the metal cluster, thereby altering the orientations of the diagonally placed metal-cluster nodes C2 and C4. In the non-interpenetrated structure all four tp units have one carboxylate coordinated to the metal centre in chelating mode whereas the second carboxylate group is coordinated in both bridging and chelating fashion. However, in the corresponding doubly-interpenetrated structure, both of the carboxylate groups of L2 and L4 are coordinated to the metal centre in chelating fashion only, whereas both of the carboxylate groups of L1 and L3 are coordinated in bridging as well as chelating mode. Furthermore, the pyridyl rings of the 4,4′-bpy ligands are twisted relative to one another with a dihedral angle of 38.7(8)°. Owing to the changes in metal cluster nodes C2 and C4, all four dinuclear Cd(II) clusters (C1 to C4) assume the same general orientation in the twofold-interpenetrated framework. During the transformation of 1 from being non-interpenetrated to doubly-interpenetrated the diagonal distances between the metal cluster centres (i.e. the Cd⋯Cd centroid) changes from 21.29(7) × 12.95(7) Å to 20.17(4) × 16.11(3) Å (ESI†). We also note that the originally planar geometry of the ring C1–C4 becomes distorted as a result of the transformation.
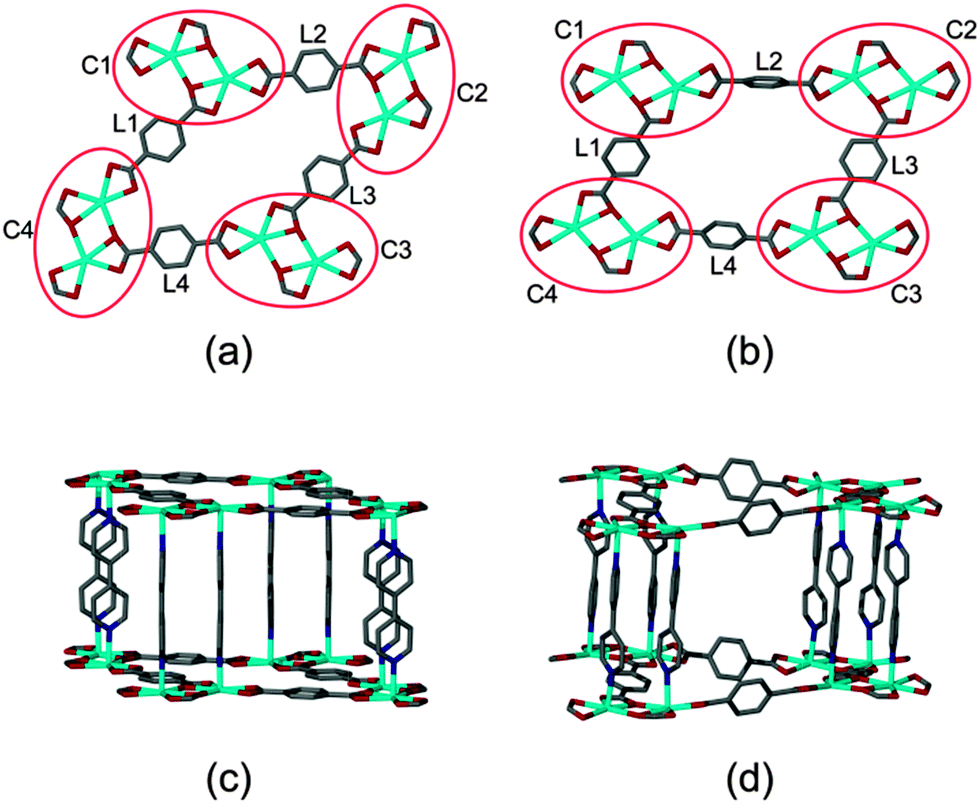 | ||
| Fig. 4 Comparison of corresponding parts of an individual network representing framework 1 in both its non-interpenetrating and doubly-interpenetrating forms: arrangement of metal clusters and tp units in the (a) non-interpenetrated and (b) doubly-interpenetrated forms and arrangement of the 4,4′-bpy ligands in the (c) non-interpenetrated and (d) doubly-interpenetrated forms. | ||
A similar rearrangement of the metal clusters and ligand coordination mode occurs during the transformation of 2 (Fig. 5). The diagonal distances between the metal clusters change from 21.10(3) × 13.70(2) Å to 20.48(6) × 15.95(2) Å on conversion from non-interpenetrated to twofold interpenetrated 2 (ESI†).
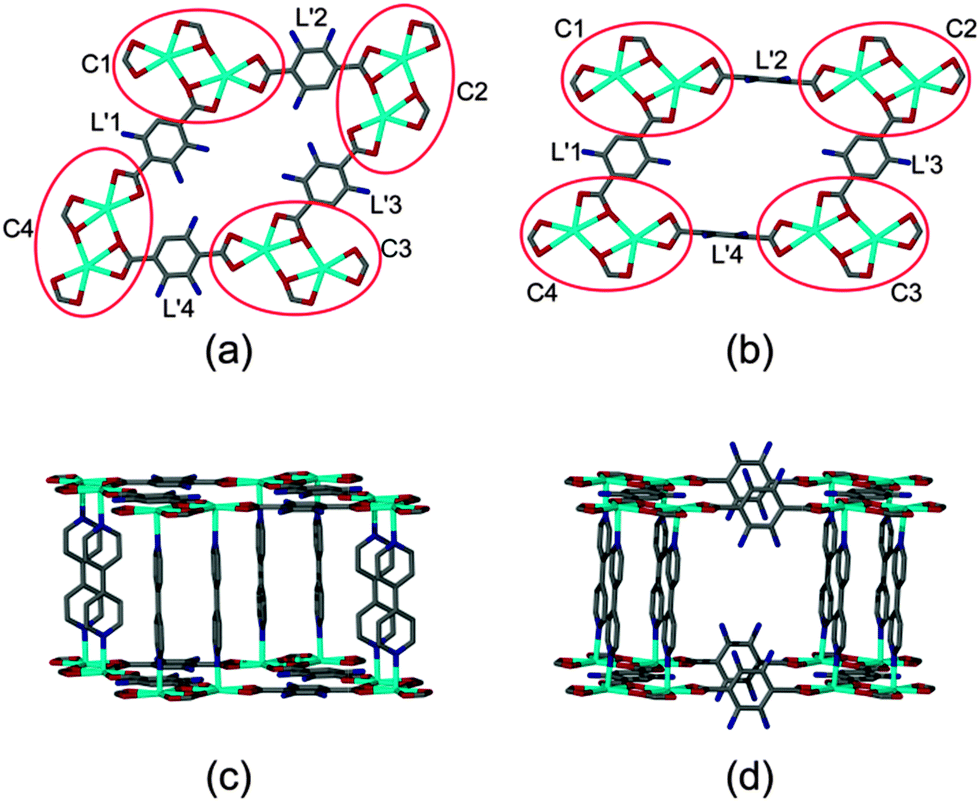 | ||
| Fig. 5 Comparison of corresponding parts of an individual network representing framework 2 in both its non-interpenetrating and doubly-interpenetrating forms: arrangement of metal clusters and atp units in the (a) non-interpenetrated and (b) doubly-interpenetrated forms and arrangement of the 4,4′-bpy ligands in the (c) non-interpenetrated and (d) doubly-interpenetrated forms. | ||
We have shown that robust doubly-pillared structures can also undergo change in degree of interpenetration upon loss of solvent molecules from the channels. The transformation occurs at 150 °C in the case of framework 1, whereas the analogous conversion of framework 2 requires a much higher temperature. This is due to the presence of the amino group in 2, which most likely obstructs the channels, thereby making it more difficult for the system to accommodate another network. Over the past few years there has been significant interest19 in the post-synthetic modification (PSM) of MOFs and, in this regard, a change in the degree of interpenetration can be considered to be a new form of PSM. We believe that similar transformations are possible in other MOF systems and that a more detailed study is required in order to better understand such phenomena.
We are grateful to the SARChI Programme of the Department of Science and Technology and the National Research Foundation (South Africa) for financial support. P. L thanks the Claude Leon Foundation for financial support.
Notes and references
- (a) L. J. Barbour, M. R. Caira, A. Coetzee and L. R. Nassimbeni, J. Chem. Soc., Perkin Trans. 2, 1995, 1345–1349 RSC; (b) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS PubMed; (c) J. L. Atwood, L. J. Barbour and A. Jerga, Chem. Commun., 2002, 2952–2953 RSC; (d) L. J. Barbour, M. R. Caira, T. le Roex and L. R. Nassimbeni, J. Chem. Soc., Perkin Trans. 2, 2002, 1973–1979 RSC; (e) L. Dobrzańska, G. O. Lloyd, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2006, 45, 5856–5859 CrossRef PubMed; (f) D. Das, E. Engel and L. J. Barbour, Chem. Commun., 2010, 46, 1676–1678 RSC; (g) T. Jacobs, J.-A. Gertenbach, D. Das and L. J. Barbour, Aust. J. Chem., 2010, 63, 573–577 CrossRef CAS; (h) M. Lusi and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed; (i) M. Lusi and L. J. Barbour, Chem. Commun., 2013, 49, 2634–2636 RSC; (j) M. Lusi and L. J. Barbour, CrystEngComm, 2014, 16, 36–38 RSC; (k) M. du Plessis, V. J. Smith and L. J. Barbour, CrystEngComm, 2014, 16, 4126–4132 RSC.
- (a) D. Bradshaw, J. E. Warren and M. J. Rosseinsky, Science, 2007, 315, 977–980 CrossRef CAS PubMed; (b) L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631–637 CrossRef CAS PubMed; (c) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC; (d) M. Kawano and M. Fujita, Coord. Chem. Rev., 2007, 251, 2592–2605 CrossRef CAS; (e) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS.
- (a) B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed; (b) D. Han, F.-L. Jiang, M.-Y. Wu, L. Chen, Q.-H. Chen and M.-C. Hong, Chem. Commun., 2011, 47, 9861–9863 RSC; (c) H. Kim, S. Das, M. G. Kim, D. N. Dybtsev, Y. Kim and K. Kim, Inorg. Chem., 2011, 50, 3691–3696 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- (a) H.-L. Jiang, T. A. Makala and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232–2249 CrossRef CAS; (b) J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed.
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS; (b) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC; (c) R. K. Das, A. Aijaz, M. K. Sharma, P. Lama and P. K. Bharadwaj, Chem. – Eur. J., 2012, 18, 6866–6872 CrossRef CAS PubMed; (d) C. Dey and R. Banerjee, Chem. Commun., 2013, 49, 6617–6619 RSC.
- (a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed; (b) T. Jacobs, G. O. Lloyd, J.-A. Gertenbach, K. K. Müller-Nedebock, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 124, 4997–5000 CrossRef.
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem. – Eur. J., 2005, 11, 3521–3529 CrossRef CAS PubMed.
- O. Shekhah, H. Wang, M. Paradinas, C. Ocal, B. Schüpbach, A. Terfort, D. Zacher, R. A. Fischer and C. Wöll, Nat. Mater., 2009, 8, 481–484 CrossRef CAS PubMed.
- H.-L. Jiang, Yo. Tatsu, Z.-H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed.
- J. Zhang, L. Wojtas, R. W. Larsen, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2009, 131, 17040–17041 CrossRef CAS PubMed.
- (a) H. M. Guo and Z.-M. Sun, J. Mater. Chem., 2012, 22, 15939–15946 RSC; (b) L. Ma and W. Lin, J. Am. Chem. Soc., 2008, 130, 13834–13835 CrossRef CAS PubMed.
- (a) L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; (b) T. K. Prasad and M. P. Suh, Chem. – Eur. J., 2012, 18, 8673–8680 CrossRef CAS PubMed.
- H. Aggarwal, P. M. Bhatt, C. X. Bezuidenhout and L. J. Barbour, J. Am. Chem. Soc., 2014, 136, 3776–3779 CrossRef CAS PubMed.
- J. Tao, M.-L. Tongand and X.-M. Chen, J. Chem. Soc., Dalton Trans., 2000, 3669–3674 RSC.
- The free volume values given are those reported in the original publications. Using Mercury ( C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS ), we have calculated guest-accessible volumes of 30.4% and 28.3% for frameworks 1 and 2, respectively.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS PubMed.
- S. B. Choi, H. Furukawa, H. J. Nam, D.-Y. Jung, Y. H. Jhon, A. Walton, D. Book, M. O’Keeffe, O. M. Yaghi and J. Kim, Angew. Chem., Int. Ed., 2012, 51, 8791–8795 CrossRef CAS PubMed.
- (a) M.-H. Zeng, Z. Yin, Y.-X. Tan, W.-X. Zhang, Y.-P. He and M. Kurmoo, J. Am. Chem. Soc., 2014, 136, 4680–4688 CrossRef CAS PubMed; (b) M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed; (c) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed; (d) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC; (e) R. K. Deshpande, J. L. Minnaar and S. G. Telfer, Angew. Chem., Int. Ed., 2010, 49, 4598–4602 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, thermal analysis, crystallographic information, Rietveld refinement parameters and additional figures. See DOI: 10.1039/c4cc07112c |
| This journal is © The Royal Society of Chemistry 2014 |