
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparation of planar rotamers through intramolecular hydrogen bonding in polysubstituted 5-nitrosopyrimidines†‡
L.
Čechová§
ab,
E.
Procházková§
ac,
I.
Císařová
c,
M.
Dračínský
*a and
Z.
Janeba
*a
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Flemingovo nám. 2, CZ-166 10 Prague 6, Czech Republic. E-mail: janeba@uochb.cas.cz; dracinsky@uochb.cas.cz
bDepartment of Chemistry of Natural Compounds, Institute of Chemical Technology Prague, Technická 5, CZ-166 28 Prague 6, Czech Republic
cFaculty of Science, Charles University in Prague, Hlavova 2030, CZ-128 40 Prague 2, Czech Republic
First published on 8th October 2014
Abstract
While purifying new polysubstituted 5-nitrosopyrimidines, the unique separation of pairs of rotamers as chemical species, stabilized by a single intramolecular hydrogen bond and differing only in nitroso group orientation, was achieved. This interesting stereochemical phenomenon is compared to the well-known atropisomerism.
The ability to form strong hydrogen bonds is one of the most important properties of biologically active compounds. Intermolecular hydrogen bonding is crucial for encoding genetic information (Watson–Crick base pairs); intramolecular hydrogen bonding is pivotal to the formation of biologically relevant three-dimensional structures of macromolecules, such as nucleic acids and proteins. Nevertheless, the ability to form intramolecular hydrogen bonds (IMHBs) also has a great impact on the molecular structure and physico-chemical properties of small organic molecules, making the closed form (with an IMHB) more lipophilic and the open form more water-soluble.1 Furthermore, the cyclic system formed due to the presence of an IMHB can mimic the structural motifs of other biologically relevant molecules. Thus, replacing the real ring with a “pseudo” ring in drug-like molecules has become an important medicinal chemistry strategy in the search for new lead compounds.1,2
The high stability of IMHBs has often been observed in planar systems where a six-membered ring is formed and the linker atoms are sp2-hybridized (as in amides, enols of β-diketones and β-enaminones, heteroaromatic rings, etc.).1 Some researchers suggest that the stability of such systems is due to stabilization by a resonance-assisted hydrogen bond (RAHB).3 Others believe that RAHB is not the primary reason behind the strength of the IMHBs in a series of enols of β-diketones and β-enaminones, but rather that the molecular structure (σ-skeleton) keeps the hydrogen donor and hydrogen acceptor coplanar and, therefore, closer to each other.4
An effect of the internal hydrogen bond on the conformational isomerism of 2-amino-2-methylpropanol was theoretically studied by Andrade et al.5 Recently, conformers of C-nitroso derivatives of indolizines and azaindolizines with restricted rotation about the C–NO bond were studied by the Katritzky group using solution NMR methods.6 Conjugated nitrosamines can evidently form planar systems with enhanced π-delocalization and, thus, belong to a class of compounds able to form strong IMHBs.7 During recent studies of 5-nitrosopyrimidines bearing distinct amino substituents in the C4 and C6 positions, two possible rotamers, stabilized by strong IMHBs, were identified as two sets of signals in 1H NMR spectra.8 In our previous paper,8d the ratio of the two rotamers was found to be significantly substituent-dependent with a broad range of conformation ratios. NMR spectroscopy showed that the interconversion barriers of these rotamers were as high (>20 kcal mol−1) as those reported for atropisomers,9 which are conformational isomers isolable through sterically hindered rotation. The high interconversion barriers of these rotamers led us to wonder: could the use of low temperatures with appropriately substituted amino groups in positions C4 and/or C6 potentially lead to the separation of a pair of IMHB-stabilized rotamers? As non-covalent interactions (e.g. hydrogen bonds) are considerably less stable compared to covalent bonds, it has been suggested by Szumna10 that separation of isomers based on a single hydrogen bond is not possible, unless additional auxiliaries are introduced. Indeed, the isolation of such isomers has so far been successful either in relatively complex molecules with an ability to form cooperative robust systems of many hydrogen bonds,11 or in the case of relatively stable formamide rotamers.12 Although spatial separation using an electric field has recently been developed to enable the study of small molecule conformers,13 the isolation and full characterization of small molecule rotamers of aromatic compounds as separate chemical species stabilized by a single resonance-assisted IMHB has not yet been reported.
In this paper, we present unique examples of the separation and full characterization of such planar rotamers isolable through a single intramolecular hydrogen bond.
Our previously described methodology8d (see ESI‡) was used to prepare a new set of polysubstituted 5-nitrosopyrimidines bearing two different hydrogen-bond donors in positions C4 and C6 of the pyrimidine moiety. While purifying compounds 1–4 (Fig. 1), two products were observed as partially separated UPLC peaks and as well-separated spots on thin layer chromatography (TLC) plates. These products were isolated by routine silica gel chromatography and, to our surprise, found to have identical mass and subsequently identified as A and B rotamer pairs (Fig. 1), differing only in their 5-nitroso group orientation. The purification of compounds 1 and 2 led to the isolation of pure rotamers 1A/1B and 2A/2B. In the case of compounds 3 and 4, it was only possible to isolate 3A and 4A as pure isomers; 3B and 4B were always, to some extent, contaminated by 3A and 4A, respectively.
 | ||
| Fig. 1 Two possible geometries of the nitroso group in position C5 for compounds 1–4. Rotamer A has a torsion angle C6–C5–N–O close to 0°; rotamer B has a torsion angle C6–C5–N–O close to 180°. | ||
We were quite astonished by the fact that we were able to separate rotamers A and B (Fig. 1) at room temperature and characterize them using just standard spectroscopic methods. Initially, we considered this phenomenon to be a special case of atropisomerism because atropisomers are, by definition,9 stereoisomers with restricted rotation around a single bond where the rotational barrier is high enough to permit isolation of the isomeric species. However, we quickly realized that there were significant differences between typical atropisomers and our rotamers, the most obvious being their mode of stabilization. Steric hindrance stabilizes atropisomers, but our planar rotamers were stabilized by intramolecular hydrogen bonding (Fig. 2).
 | ||
| Fig. 2 General characteristics of atropisomerism and the herein reported stereochemical phenomenon. A ≠ B and A′ ≠ B′, A/A′ has priority over B/B′. | ||
Having made this distinction, we explored further similarities and differences between atropisomers and our planar rotamers. We found that there are two necessary preconditions for both the phenomena. The first precondition is a rotationally stable axis, which, in our case, lies along the C5–NO bond. Analogous to the axis of the most common class of atropisomers represented by biaryl compounds (the sp2–sp2 family),9 the herein reported rotamers are characterized by a rotationally stable bond between sp2 carbon (C5) and sp2 nitrogen (nitroso group) atoms. The second precondition involves the presence of different substituents on both sides of the axis; in our case, there are differently substituted amino groups in the C4 and C6 positions, and an oxygen and free electron pair on the nitrosyl nitrogen. However, unlike atropisomers,9 which contain an element of chirality (usually through the axis), our planar rotamers (shortly planamers), precisely because they are planar, lack any element of chirality.
The solid-state 13C cross polarization-magic angle spinning (CP-MAS) NMR spectra of rotamers 1A and 1B obtained after gentle evaporation of the solvents from the chromatographic fractions are depicted in Fig. 3 and Fig. S1 (ESI‡). According to these spectra, both rotamers were pure; the amount of the second one in the pair was lower than the solid-state NMR detection limit (ca. 5%). The 13C NMR signals of rotamer 1A are sharp, indicating long-range order in the solid, while the spectrum of rotamer 1B exhibits the features typical of amorphous structures; namely, broad signals with multiple overlapped resonances. Interestingly, the solid-state 13C NMR spectra show that while the crystalline rotamer 1A was perfectly stable at room temperature, the solid amorphous rotamer 1B slowly re-crystallized, over a period of months, into the crystalline rotamer 1A (Fig. 3 and Fig. S1 in ESI‡).
 | ||
| Fig. 3 The aromatic region of 13C CP-MAS solid-state NMR spectra of planar rotamers 1A and 1B. Solid amorphous 1B slowly transforms into solid crystalline 1A. The full spectrum is available in Fig. S1 in ESI.‡ | ||
The solid-state IR spectra of rotamers 1A and 1B were acquired on samples dispersed in KBr pellets. Similar to the NMR spectra, there were significant differences between the rotamers. For example, a difference of up to 40 cm−1 was observed between their antisymmetric N–H vibrations. Furthermore, as with the NMR spectra, the signals of the amorphous isomer 1B were slightly broader than those of the crystalline isomer 1A, with no visible contamination by each other (Fig. S2 in ESI‡).
After the re-crystallization of compound 1 (a mixture of 1A and 1B) from dry acetone, a monocrystal suitable for X-ray diffraction (XRD) was obtained (Fig. 4A). XRD confirmed that the orientation of the nitroso group corresponded to that of pure rotamer 1A. The solid-state 13C NMR spectrum of this recrystallized material was identical to the spectrum of 1A obtained by column chromatography. However, following re-crystallization from an acetone–water mixture, a monohydrate of compound 1 was obtained. In this monohydrate (Fig. 4B), both rotamers were present, 1A being predominant (ca. 80%). Clearly, in a solid state, the conformational stability of a rotamer depends on the polymorphic/crystalline form of the parent compound. In this way, our planamers meet Oki's criterion for atropisomers:14 physically separable species at a given temperature, having a half-life (τ) of at least 1000 s (∼16.7 min), which is the time considered to be the minimum lifetime for a molecule to be isolable.
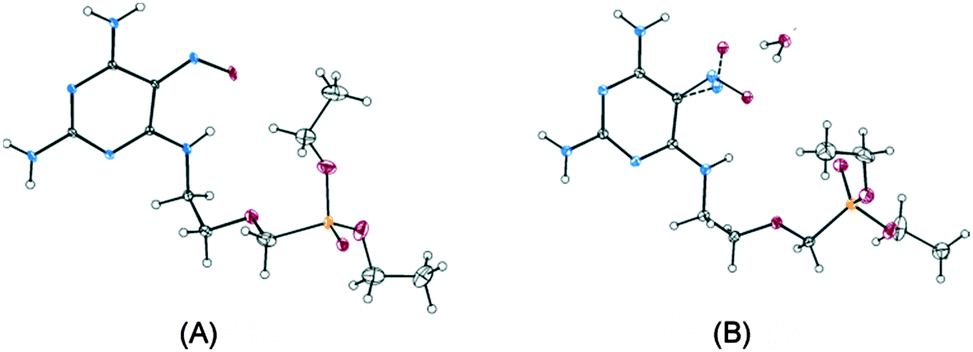 | ||
| Fig. 4 X-ray crystallographic analysis of pure rotamer 1A (A) and of the monohydrate of compound 1 (B). Carbon is shown in grey, nitrogen in blue, oxygen in red and phosphorus in yellow. The less occupied position of the nitroso group of rotamer 1B is drawn with dashed lines (B). The displacement ellipsoids are drawn at a 30% probability level. The disordered atoms of the phosphonate moiety and solvating water molecule are omitted for clarity. | ||
Immediately after dissolution of the separated rotamers A or B in DMSO-d6 or CD3OD, their 1H and 13C NMR spectra contained one major set of signals together with the low intensity signals of its corresponding second rotamer, B or A, respectively. Gradually, the A/B signal ratio changed until, after several hours, equilibrium was reached. The kinetics of these transformations were determined by solution-state 1H NMR spectroscopy (Fig. 5 and Fig. S3 in ESI‡). The NH hydrogen signals of the two rotamers were usually well separated and, thus, their integration enabled the mixture composition to be determined at various times after dissolution. Table S1 (see ESI‡) summarizes the rate constants (calculated according to the procedure in ESI‡) and the concentrations of rotamers 1A–4A in equilibrium in various solvents. It is evident that the rotamer equilibrium ratio was not only strongly substituent dependent, but also solvent dependent.
 | ||
| Fig. 5 Interconversion of rotamers 1A and 1B. Interconversion was monitored by 1H NMR spectroscopy in a DMSO-d6 solution. R1 = CH2–CH2–O–CH2–P(O)(OEt)2. | ||
The signals of the NH hydrogens involved in the hydrogen bond were shifted downfield (10–13 ppm) with respect to the signals of the free NH hydrogens (7–9 ppm). The position and orientation of the hydrogen-bond donor atom was confirmed both by the splitting of the signal, which resulted from indirect spin–spin interactions with neighbouring hydrogen atoms, and by the two- and three-bond correlations observed in the HMBC spectra. We also observed four-bond correlations between the NH hydrogen atoms and the pyrimidine carbon atoms. These correlations were observed when the atoms involved were arranged in the form of a W-like coupling path (Fig. S4 in ESI‡). Furthermore, the orientation of the nitroso group had a significant influence on the chemical shifts of C4 and C6: by ca. 12 ppm, the C6 carbon was more shielded in rotamer A and the C4 carbon more shielded in rotamer B.
Fig. 6 shows the progress of the isotope exchange of hydrogen C6–NH for compound 1 in a DMSO-d6–CD3OD mixture. As can be seen from the rates of the hydrogen-to-deuterium exchange reaction, the formation of intramolecular hydrogen bonds reduced the accessibility of the solvent to the hydrogen bond donors. In rotamer 1A, where C6–NH is involved in the hydrogen bond, the exchange reaction was much slower than in rotamer 1B. As is normal for free amino groups, the hydrogen atoms of the amino group in position C2 were completely replaced by deuterium before the first spectrum could be acquired. Interestingly, both amino hydrogens in position C4 in isomer 1B were also fully replaced before the first data acquisition. We assume that this was caused by the fast isotope exchange of the unbound amino hydrogen followed by fast rotation around the C4–NH2 bond. This hypothesis is supported by the density functional theory (DFT) calculations for the C4–NH2 rotation barrier, which was found to be 5.6 kcal mol−1 lower than the rotation of the nitroso group around the C5–NO bond (see ESI‡).
 | ||
Fig. 6 The progress of the H–D exchange of hydrogen atoms of the amino group in position C6 in the equilibrium mixture of rotamers 1A and 1B in a DMSO-d6–CD3OD mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1). | ||
In summary, we succeeded in preparation of planar rotamers which, like atropisomers, are characterized by a high interconversion barrier that enables their separation by column chromatography at room temperature. Herein, we have isolated and fully characterized planar rotamers structurally derived from polysubstituted 5-nitrosopyrimidines. Unlike atropisomers, whose separation is achieved through steric hindrance (and display chirality), the isolation of our planar rotamers is achieved through intramolecular hydrogen bonding (and are achiral). To make a distinction from relatively common atropisomerism, we suggest a term ‘planamerism’ and define ‘planamers’ as small aromatic molecule rotamers with a planar conjugated moiety that are isolable as chemical species. It is possible that the reported stereochemical phenomenon has a role to play in disciplines involving weak and reversible non-covalent interactions, such as supramolecular assemblies, host molecules, or in the development of novel drug-like molecules. Therefore, to more clearly understand the scale and scope of this phenomenon, we are currently synthesizing other substituted (hetero)aromatic derivatives likely to be capable of forming strong intramolecular hydrogen bonds and, consequently, separable rotamers.
The study is a part of the Research Project RVO61388963 of the Institute of Organic Chemistry and Biochemistry, the Academy of Sciences of the Czech Republic, v.v.i., and was supported by the Ministry of Interior of the Czech Republic (VG20102015046), the Czech Science Foundation (grant no. 13-24880S) and Gilead Sciences (Foster City, CA, USA). We acknowledge Dr Lucie Bednárová and Pavel Fiedler (Molecular Spectroscopy, Institute of Organic Chemistry and Biochemistry) for the measurement of the IR spectra.
Notes and references
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601 CrossRef CAS PubMed.
- (a) C. N. Hodge and J. Pierce, Bioorg. Med. Chem. Lett., 1993, 3, 1605 CrossRef CAS; (b) P. Furet, G. Bold, F. Hofmann, P. Manley, T. Meyer and K.-H. Altmann, Bioorg. Med. Chem. Lett., 2003, 13, 2967 CrossRef CAS PubMed; (c) P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer, Bioorg. Med. Chem. Lett., 2008, 18, 897 CrossRef CAS PubMed; (d) K. A. Menear, C. Adcock, F. C. Alonso, K. Blackburn, L. Copsey, J. Drzewiecki, A. Fundo, A. Le Gall, S. Gomez, H. Javaid, C. F. Lence, N. M. B. Martin, C. Mydlowski and G. C. M. Smith, Bioorg. Med. Chem. Lett., 2008, 18, 3942 CrossRef CAS PubMed; (e) A.-M. Lord, M. F. Mahon, M. D. Lloyd and M. D. Threadgill, J. Med. Chem., 2009, 52, 868 CrossRef CAS PubMed.
- (a) G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023 CrossRef CAS; (b) S. Bolvig and P. E. Hansen, Curr. Org. Chem., 2000, 4, 19 CrossRef CAS; (c) L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513 CrossRef CAS PubMed; (d) K. A. Lyssenko and M. Y. Antipin, Russ. Chem. Bull., 2006, 55, 1 CrossRef CAS.
- P. Sanz, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. A, 2007, 111, 3585 CrossRef CAS PubMed.
- L. A. F. Andrade, J. M. Silla and M. P. Freitas, J. Mol. Struct., 2014, 1072, 203 CrossRef CAS.
- I. Ghiviriga, B. E.-D. M. El-Gendy, H. Martinez, D. Fedoseyenko, E. P. Metais, A. Fadli and A. R. Katritzky, Org. Biomol. Chem., 2010, 8, 3518 CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef CAS.
- (a) I. Susvilo, A. Brukstus and S. Tumkevicius, Tetrahedron Lett., 2005, 46, 1841 CrossRef CAS; (b) G. Urbelis, I. Susvilo and S. Tumkevicius, J. Mol. Model., 2007, 13, 219 CrossRef CAS PubMed; (c) A. Marchal, M. Nogueras, A. Sánchez, J. N. Low, L. Naesens, E. De Clercq and M. Melguizo, Eur. J. Org. Chem., 2010, 3823 CrossRef CAS; (d) E. Procházková, L. Čechová, Z. Janeba and M. Dračínský, J. Org. Chem., 2013, 78, 10121 CrossRef PubMed.
- (a) E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994 Search PubMed; (b) I. Alkorta, J. Elguero, C. Roussel, N. Vanthuyne and P. Piras, Adv. Heterocycl. Chem., 2012, 105, 1 CrossRef CAS.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274 RSC.
- K. Mishiro, T. Furuta, T. Sasamori, K. Hayashi, N. Tokitoh, S. Futaki and T. Kawabata, J. Am. Chem. Soc., 2013, 135, 13644 CrossRef CAS PubMed.
- M. Geffe, L. Andernach, O. Trapp and T. Opatz, Beilstein J. Org. Chem., 2014, 10, 701 CrossRef CAS PubMed.
- (a) F. Filsinger, J. Küpper, G. Meijer, J. L. Hansen, J. Maurer, J. H. Nielsen, L. Holmegaard and H. Stapelfeldt, Angew. Chem., Int. Ed., 2009, 48, 6900 CrossRef CAS PubMed; (b) T. S. Zwier, Nat. Chem., 2009, 1, 687 CrossRef CAS PubMed.
- K. Oki, Recent Advances in Atropisomerism, Topics in Stereochemistry, Wiley Interscience, New York, 1983, vol. 14, pp. 1–76 Search PubMed.
Footnotes |
| † This paper is dedicated to the memory of Professor Alan R. Katritzky, who died on February 10, 2014. |
| ‡ Electronic supplementary information (ESI) available: Supplementary figures and Table, X-ray diffraction data, kinetics, DFT calculations, synthesis and characterization of the prepared compounds. CCDC 1001296 and 1001297. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc06978a |
| § L.C. and E.P. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |