
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile single crystal to single crystal transition with significant structural contraction on desolvation†
Jane V.
Knichal
a,
William J.
Gee
a,
Andrew D.
Burrows
*a,
Paul R.
Raithby
*a,
Simon J.
Teat
b and
Chick C.
Wilson
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: C.C.Wilson@bath.ac.uk; Web: http://www.metastable-materials.org.uk/
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 947240, USA
First published on 1st October 2014
Abstract
5-Ethynyl-1,3-benzenedicarboxylic acid (H2ebdc) reacted with lead(II) acetate trihydrate yields a 1D ladder network, [Pb(ebdc)(MeOH)]2·H2O (1). Removing crystals of 1 from the mother liquor results in a facile single crystal to single crystal transition, yielding 2D [Pb(ebdc)] net (2) with a change in space group from I2/a to P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .
.
Single crystal to single crystal (SCSC) transitions are a fascinating post-synthetic solid-state class of reactions with important implications that range from the manipulation of pharmaceutical properties,1 to understanding the pathological effects of disease2 to modulating molecular magnetism.3 The scope of SCSC transitions include migration of guest molecules, changes to ligand conformation, and variations of the metal coordination sphere.4 Examples involving addition of guest molecules,5 loss of guest molecules,4,5b,c,6 post-synthetic modification,7 inducement by variation of pressure,8 temperature variation,9 and photoexcitation10 are known, however the scarcity of these systems prevent detailed study of SCSC transitions. Two reviews discussing the solid-state reactivity and structural transformations have recently appeared.11 Serendipity has dominated discovery of SCSC systems, although our efforts in characterizing metastable materials are an attempt to address this.12
Two SCSC transitions involving lead coordination polymers (PbCPs) are known that require either elevated temperatures (≥160 °C, 3 days)13 or photoirradiation (60 hours) to effect.14 The latter transformation was described as an example of drastic molecular motions,11b however it occurred with concomitant loss of single crystallinity. The work described here exceeds the degree of molecular motion observed in past PbCPs while retaining single crystallinity, allowing detailed solid-state analysis.
The compound [Pb(ebdc)(MeOH)]2·H2O (1) crystallises in the monoclinic space group I2/a and has an asymmetric unit that contains a divalent lead centre coordinated to a single 5-ethynyl-1,3-benzenedicarboxylate (ebdc) ligand, and a molecule of methanol. The ebdc ligand chelates to the lead(II) centre via one carboxylate group in a κO:κO′ manner, and bridges via the second carboxylate in a μ-(κO:κO′) fashion to two symmetry generated (−x + 1/2, y, −z + 1 and x − 1, y, z) lead(II) atoms. Accounting for symmetry, the lead(II) atom possesses five primary bonds (<2.75 Å)15 the sum of which yield a 1D ladder network that propagates along the crystallographic a axis. There are two additional secondary interactions within the limits of van der Waals' interactions for lead and oxygen (2.75 < 3.30 Å), O(3′)–Pb(1): 2.876(6) and O(4′′)–Pb(1): 2.837(5) Å. Taken into account, these weaker interactions give a 2D network (Fig. 1). We believe the distinction between secondary interactions and primary bonds is important, as the making or breaking of these bonds appears to facilitate the SCSC process (vide infra).‡
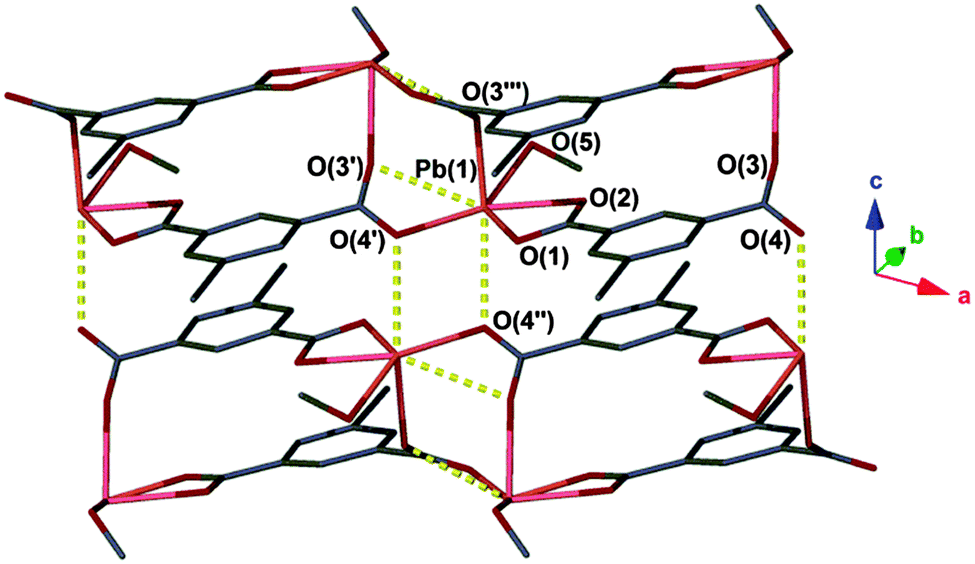 | ||
| Fig. 1 Coordination environment of 1. Secondary interactions are represented as yellow dashed lines. Hydrogen atoms and water molecules have been omitted for clarity. Unit cell: a = 10.037(2) Å, b = 17.698(4) Å, c = 12.687(3) Å, β = 96.75(3)°. | ||
Alignment of the ethynyl groups along the network provides ordering in the solid state by interdigitation, such that opposing ladders associate in a zipper-like fashion. Weak hydrogen bonding deriving from the ethynyl groups was observed, and protonation of the group was confirmed by the C–H ethynyl band at 3240 cm−1 in the IR spectrum.
Lattice water molecules sit between adjacent chains of 1 acting as bifurcated hydrogen bond acceptors from two coordinated methanol molecules (O(5)⋯O(6): 2.725(10) Å), and donors to two carboxylate moieties (O(6)⋯O(2): 2.749(8) Å). These hydrogen bonding regions separate interdigitated pairs of ladders ordering the structure (Fig. 2).
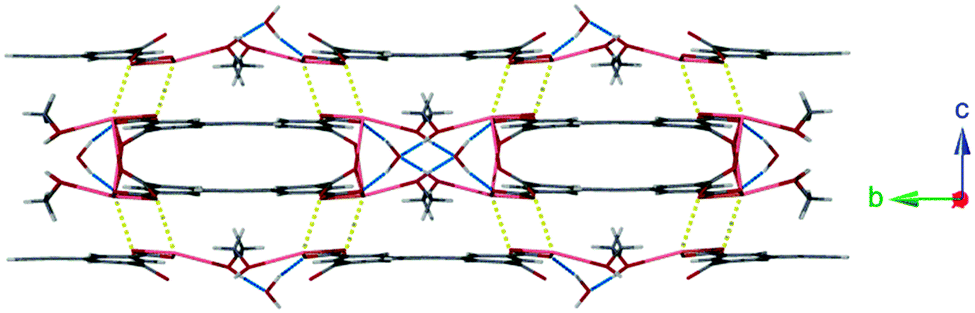 | ||
| Fig. 2 Hydrogen bonding interactions between 1D ladders of 1 (shown in blue). Also highlighted are weaker secondary Pb–O interactions (shown in yellow). | ||
Exposing a crystalline powder of 1 to air over the course of several hours resulted in conversion to a second species, [Pb(ebdc)] (2), which adopts the triclinic space group P, and contains two crystallographically distinct lead(II) centres. The lead(II) atoms are bridged by two dicarboxylate ligands within the asymmetric unit. Symmetry between the lead centres is broken by formation of a new primary bond from Pb(1) to O(1′′) with a distance of 2.719(8) Å. Consequently, the sum of the primary coordination interactions yields an unusual four-legged ladder motif (Fig. 3). The loss of coordinating solvent and lattice water during the SCSC transition replaces the hydrogen bonding interactions observed for 1 with two new secondary interactions, O(3′′)–Pb(2): 2.989(7) and Pb(1)–O(6′′): 2.817(7) Å (Fig. 4, right; Fig. S6, ESI†).
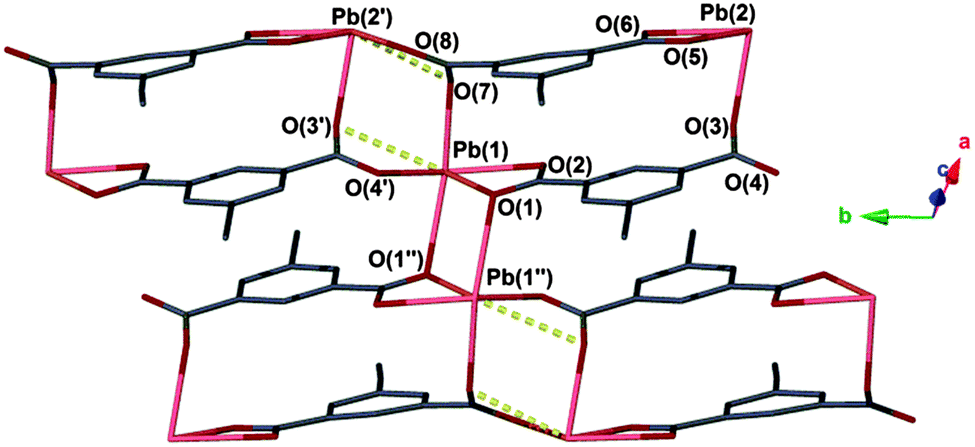 | ||
| Fig. 3 Coordination environment of 2. Secondary interactions are represented as yellow dashed lines. Hydrogen atoms are omitted for clarity. Unit cell: a = 9.3790(4) Å, b = 10.0130(3) Å, c = 10.3180(4) Å, α = 100.148(3)°, β = 91.407(3)°, γ = 107.941(3)°. | ||
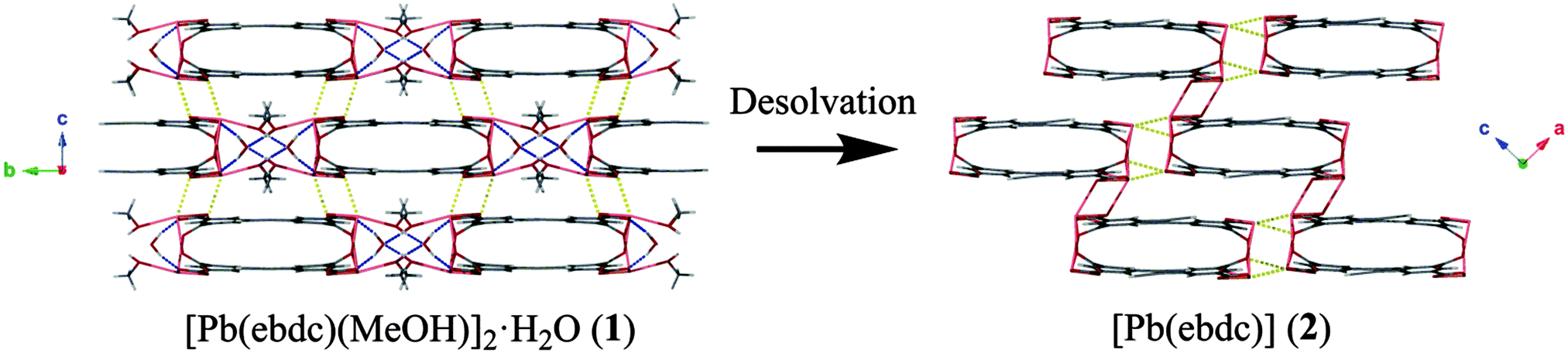 | ||
| Fig. 4 Loss of solvated regions of the crystal, viewed here down the crystallographic a axis of 1 and b axis of 2, cause the 1D ladders to compress, reducing this axis by 20%. Blue dashed lines: hydrogen bonds; yellow dashed lines: secondary Pb–O interactions. | ||
Based on the crystallographic evidence, a mechanism for the SCSC transition can be suggested. Viewed down the crystallographic b and c axes respectively (Fig. 1 → Fig. 3), adjacent 1D chains of 2 have slipped in opposing directions as a result of desolvation. This has brought Pb(1) and O(1′′) into closer contact (2.719(8) Å) where formerly a distance of more than 4.00 Å was observed in 1. More dramatic changes occur along the crystallographic a axis in 1, where desolvation effects elimination of the hydrogen-bonded solvent channels and causes the interdigitated pairs of 1D polymers to converge (Fig. 4). Centroid to centroid distances measured from the centre of adjacent interdigitated pairs show a contraction along this axis of 17.7 Å for 1 to 14.1 Å for 2, equivalent to more than 20%. New secondary interactions form that stabilise 2, namely Pb(1)–O(6′′): 2.817(7) Å (1 − x, 1 − y, −z). Orthogonal to this contraction, the layers above and below shift from alternating in 1, to askew in 2, making possible the new Pb(1)–O(1′′) bond.
An unusual aspect of this SCSC transition is the facile nature of coordinated solvent molecule loss. Structural transformations involving coordinated solvent loss typically require heating to relatively high temperatures to affect conversion.16 Crystallinity of both forms was retained as determined by powder X-ray diffraction (Fig. S2, ESI†). Slow heating of a single crystal of 1 successfully yielded transition to 2 as judged by powder diffraction, however the transformation occurred with considerable degradation of the crystal above (Fig. S3, ESI†). Single crystals of 1 were next placed in an enclosed environment with limited air diffusion and allowed to stand for three days. Gratifyingly, while some degradation of the larger crystals in the sample to microcrystalline particulates was observed, small single crystals of 2 were obtained with suitable quality for X-ray analysis using a synchrotron source. The PXRD pattern of these crystals was found to match that of the degraded crystal samples. Similar relationships between crystal size and SCSC transition effectiveness have been reported for other systems.17 Exposing desolvated crystals of 2 to mixtures of methanol and water failed to recover solvated 1, demonstrating the transformation to be irreversible. The transformation from network 1 to 2 was monitored using infrared spectroscopy. The initial solvated form was readily apparent from a broad O–H band at 3297 cm−1 coupled with a methanolic C–O vibration at 1016 cm−1 deriving from the coordinated methanol. Bands at 1494 cm−1 and 1362 cm−1 correspond to respective carboxylate antisymmetric and symmetric COO− stretches. Transformation to desolvated form 2 sees the disappearance of the alcoholic bands, as well as a blue shift of the antisymmetric carboxylate stretching mode to 1516 cm−1 and a red shift in the symmetric stretching mode to 1343 cm−1, which are likely associated with formation of the new primary Pb(1)–O(7′′) bond and loss of the hydrogen bond originated from the lattice water molecule in 1. These changes occur at a single carboxylate group, which contains O(1) and O(2) in Fig. 1 and 3. The crystalline sample required 45 minutes to completely convert to the desolvated form as gauged by IR spectroscopy, a result that was supported by X-ray powder diffraction measurements.
The facile nature of the transition from 1 to 2 prompted further investigation of the behaviour within the solvent channels of 1. Immersing crystals of 1 in ethanol for several days yielded a second SCSC transition to an ethanol adduct, [Pb2(ebdc)2(EtOH)]2·H2O (3). Crystallographic analysis of 3 revealed the new crystal retains monoclinic symmetry with conversion to the I2 space group. Two crystallographically unique lead atoms and two ebdc ligands are present in the asymmetric unit, with a single ethanol molecule coordinating to Pb(1). Network 3 differs from 1 in terms of degree of solvation within the pore, and by substitution of a secondary interaction for a primary bond, which gives a 2D network motif comprised of primary bonds (Fig. 5). Replacement of coordinated methanol with a larger ethanol molecule serves to sterically displace all solvent excepting a water molecule, yielding a system with only half the hydrogen bonding interactions propagating along the crystallographic b axis as observed in 1 (Fig. 6). The single lattice water molecule again acts as a bifurcated hydrogen bond acceptor from a pair of coordinated ethanol molecules (O(9)⋯O(10): 2.732(12) Å), and donor to two carboxylate moieties (O(10)⋯O(8): 2.756(11) Å). The shortening of the secondary interaction between Pb and O(4) from 2.837(5) Å in 1, to 2.623(10) Å in 3, is likely a result of a small slip of the chains aligned to the crystallographic c axis, which allows the ethanol molecules to reorient and hydrogen bond to the lattice water molecule. The length of the crystallographic b axis for 3 remains equivalent to 1 at 17.7 Å. The conversion of 1 to 3 was found to be irreversible, and furthermore the presence of EtOH inhibits the SCSC transition of 3 to desolvated form 2 at room temperature. Heating a powder sample of 3 at 100 °C for 1 hour effected transformation to an amorphous compound that was identified as compound 2 using infrared spectroscopy. It is likely that the 2D network, which is comprised of primary bonds only in 3, provides greater stability to the hydrogen bonded solvent within the terminal alkyne bordered channels and inhibits the SCSC transition at low temperature. The transition in crystal forms derived from 1 is summarized in Scheme 1.
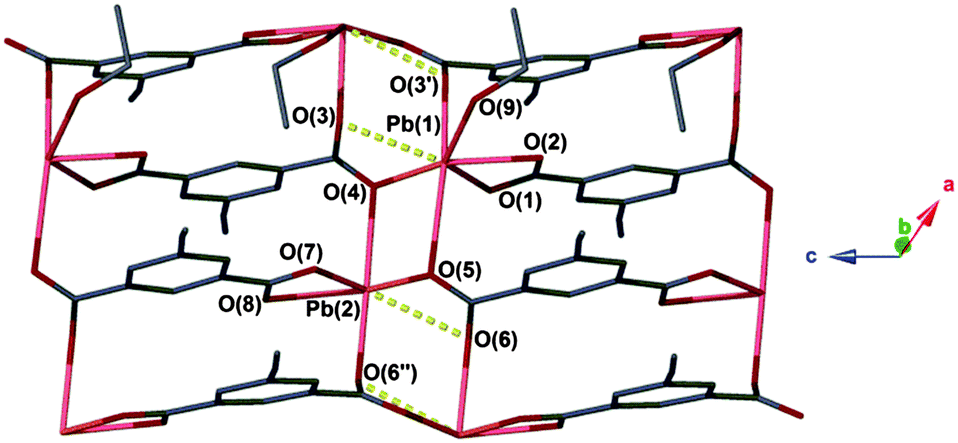 | ||
| Fig. 5 Coordination environment of 3. Secondary interactions are represented as yellow dashed lines. Hydrogen atoms are omitted for clarity. Unit cell: a = 9.988(2) Å, b = 17.698(4) Å, c = 12.489(3) Å, β = 95.59(3)°. | ||
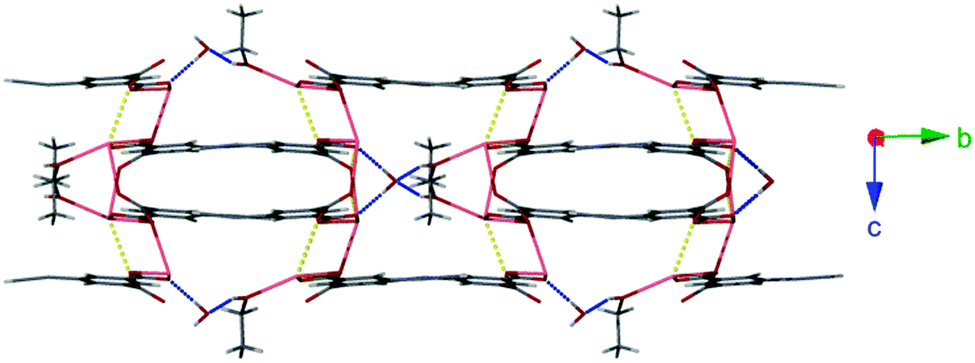 | ||
| Fig. 6 Hydrogen bonding interactions within the 2D network of 3 (blue). Also highlighted are weaker secondary coordination sphere Pb–O interactions (yellow). | ||
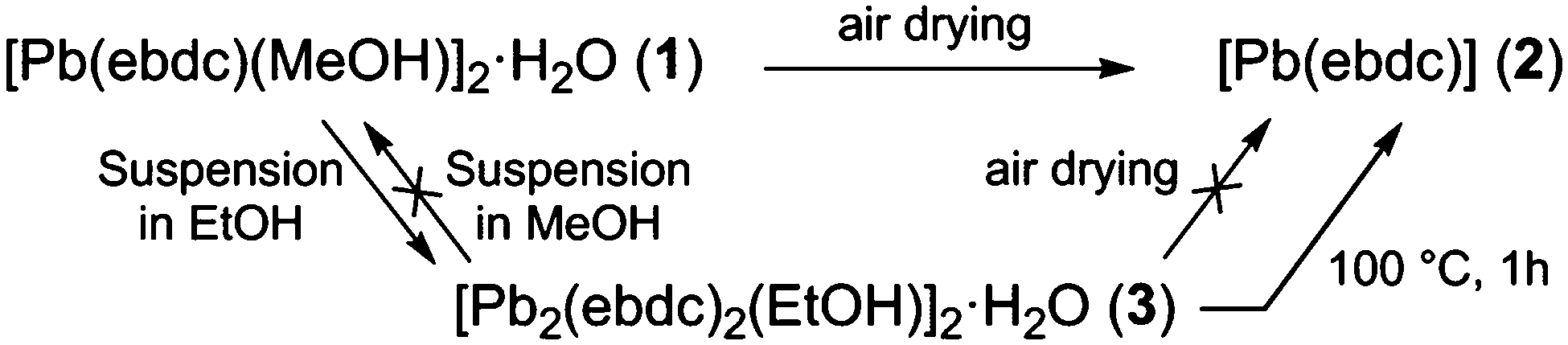 | ||
| Scheme 1 Summary of the SCSC transitions described in this work. | ||
The structures of 1–3 differ from the known lead-1,3-benzenedicarboxylate (mbdc) structures,18 which consist of three-dimensional networks. Steric effects relating to the presence of the ethynyl group in ebdc prevents the compounds formed from this ligand being isostructural to those with mbdc. The crystal data for compounds 1–3, as well as full tables of bond lengths and angles for each network, can be found in the ESI† (Tables S4 and S5).
We have identified an unusual SCSC system that undergoes large changes to crystalline topology and packing, and yet occurs via facile desolvation at room temperature, despite the requirement for coordinated solvent loss. Ease of transformation from a solvated (1 and 3) to the desolvated form (2) was found to be influenced by the nature of the Pb–O interactions. In compound 1 weak secondary interactions dominate, allowing facile slippage of the 1D chains to yield primary interactions in 2 that possess greater stability. By contrast, additional primary Pb–O interactions are present in compound 3, inhibiting this mechanism and necessitating heating to liberate the solvent and yield 2.
We are grateful to the EPSRC for financial support of the project (EP/K004956/1) and the University of Bath for a studentship to JVK. We would also like to thank the ALS, LBNL for the beamtime to perform these measurements. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Notes and references
- (a) F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, CrystEngComm, 2005, 7(29), 179 RSC; (b) F. P. A. Fabbiani, D. R. Allan, W. I. F. David, A. J. Davidson, A. R. Lennie, S. Parsons, C. R. Pulham and J. E. Warren, Cryst. Growth Des., 2007, 7, 1115 CrossRef CAS.
- I. Solomonov, M. J. Weygand, K. Kjaer, H. Rapaport and L. Leiserowitz, Biophys. J., 2005, 88, 1809 CrossRef CAS PubMed.
- (a) Y. Zhu, F. Luo, M.-B. Luo, X.-F. Feng, S. R. Batten, G.-M. Sun, S.-J. Liu and W.-Y. Xu, Dalton Trans., 2013, 42, 8545 RSC; (b) Y.-M. Song, F. Luo, M.-B. Luo, Z.-W. Liao, G.-M. Sun, X.-Z. Tian, Y. Zhu, Z.-J. Yuan, S.-J. Liu, W.-Y. Xu and X.-F. Feng, Chem. Commun., 2012, 48, 1006 RSC.
- H.-L. Sun, D.-D. Yin, Q. Chen and Z. Wang, Inorg. Chem., 2013, 52, 3582 CrossRef CAS PubMed.
- (a) T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 6666 CrossRef CAS PubMed; (b) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS PubMed; (c) G. S. Ananchenko, K. A. Udachin, A. Dubes, J. A. Ripmeester, T. Perrier and A. W. Coleman, Angew. Chem., Int. Ed., 2006, 45, 1585 CrossRef CAS PubMed.
- D. Swain and T. N. Guru Row, Inorg. Chem., 2009, 48, 7048 CrossRef CAS PubMed.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482 CrossRef CAS PubMed.
- (a) B. A. Zakharov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, B69, 271 Search PubMed; (b) M. Kikuchi, K. Hiraga, Y. Syono and H. Takei, J. Solid State Chem., 1985, 56, 390 CrossRef CAS; (c) F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, CrystEngComm, 2005, 7(29), 179 RSC; (d) K. Kusaba, T. Yagi, J. Yamaura, N. Miyajima and T. Kikegawa, Chem. Phys. Lett., 2007, 437, 61 CrossRef CAS; (e) F. P. A. Fabbiani, D. R. Allan, W. I. F. David, A. J. Davidson, A. R. Lennie, S. Parsons, C. R. Pulham and J. E. Warren, Cryst. Growth Des., 2007, 7, 1115 CrossRef CAS; (f) G. Liu, J. Liu, Y. Liu and X. Tao, J. Am. Chem. Soc., 2014, 136, 590 CrossRef CAS PubMed.
- (a) I. Agmon and M. Kaftory, J. Appl. Crystallogr., 1994, 27, 146 CrossRef CAS; (b) M. R. Caira, A. Foppoli, M. E. Sangalli, L. Zema and F. Giordano, J. Therm. Anal. Calorim., 2004, 77, 653 CrossRef CAS.
- (a) N. Nishizawa, D. Furukawa, S. Kobatake and A. Matsumoto, Cryst. Growth Des., 2010, 10, 3203 CrossRef CAS; (b) G. K. Kole, T. Kojima, M. Kawano and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 2143 CrossRef CAS PubMed.
- (a) J.-P. Zhang, P.-Q. Liao, H.-L. Zhou, R.-B. Lin and X.-M. Chen, Chem. Soc. Rev., 2014, 43, 5789 RSC; (b) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC.
- L. E. Hatcher, J. Christensen, M. L. Hamilton, J. Trincao, D. R. Allan, M. R. Warren, I. P. Clarke, M. Towrie, S. Fuertes, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 3128 CrossRef CAS PubMed.
- A. Aslani and A. Morsali, Chem. Commun., 2008, 3402 RSC.
- A. M. P. Peedikakkal and J. J. Vittal, Inorg. Chem., 2010, 49, 10 CrossRef CAS PubMed.
- R. L. Davidovich, V. Stavila, D. V. Marinin, E. I. Voit and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef CAS.
- (a) A. Neels, Y. Wang and H. Stoeckli-Evans, Z. Kristallogr., 2004, 219, 892 CAS; (b) K. Müller-Buschbaum and Y. Z. Mokaddem, Z. Anorg. Allg. Chem., 2008, 634, 2360 CrossRef; (c) A. S. R. Chesman, D. R. Turner, D. J. Price, B. Moubaraki, K. S. Murray, G. B. Deacon and S. R. Batten, Chem. Commun., 2007, 3541 RSC.
- I. Halasz, Cryst. Growth Des., 2010, 10, 2817–2823 CAS.
- (a) E.-C. Yang, J. Li, B. Ding, Q.-Q. Liang, X.-G. Wang and X.-J. Zhao, CrystEngComm, 2008, 10, 158 RSC; (b) L. Zhang, Y.-Y. Qin, Z.-J. Li, Q.-P. Lin, J.-K. Cheng, J. Zhang and Y.-G. Yao, Inorg. Chem., 2008, 47, 8286 CrossRef CAS PubMed; (c) Z. Zhang, Y.-L. Zhou and H.-Y. He, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2591 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental methods, tables of bond lengths and angles, PXRD patterns for 1–3, and TGA analysis of 1. CCDC 1007986–1007988. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc06587e |
‡ Crystal data from single crystal diffraction studies for 1. C22H18O11Pb2, Mr = 872.74, monoclinic, space group I2/c, a = 10.037(2), b = 17.698(4), c = 12.687(3) Å, β = 96.75(3)°, V = 2238.1(3) Å3, Z = 4, ρcalc = 2.590 g cm−3, total reflections = 6767, unique reflections = 2606, R1 = 0.0406, wR2 = 0.0875, GOF = 1.008, temp. = 100(2) K. For 2. C20H8O8Pb2, Mr = 790.64, triclinic, space group P, a = 9.3790(4) Å, b = 10.0130(3) Å, c = 10.3180(4) Å, α = 100.148(3)°, β = 91.407(3)°, γ = 107.941(3)°, V = 904.22(6) Å3, Z = 2, ρcalc = 2.904 g cm−3, total reflections = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 225, unique reflections = 5621, R1 = 0.0505, wR2 = 0.1064, GOF = 1.016, temp. = 100(2) K. For 3. C44H30O19Pb4, Mr = 1691.44, monoclinic, space group I2, a = 9.988(2) Å, b = 17.698(4) Å, c = 12.489(3) Å, β = 95.59(3)°, V = 2197.1(8) Å3, Z = 2, ρcalc = 2.557 g cm−3, total reflections = 24817, unique reflections = 5531, R1 = 0.0479, wR2 = 0.0908, GOF = 1.031, temp. = 149.9(2) K. 225, unique reflections = 5621, R1 = 0.0505, wR2 = 0.1064, GOF = 1.016, temp. = 100(2) K. For 3. C44H30O19Pb4, Mr = 1691.44, monoclinic, space group I2, a = 9.988(2) Å, b = 17.698(4) Å, c = 12.489(3) Å, β = 95.59(3)°, V = 2197.1(8) Å3, Z = 2, ρcalc = 2.557 g cm−3, total reflections = 24817, unique reflections = 5531, R1 = 0.0479, wR2 = 0.0908, GOF = 1.031, temp. = 149.9(2) K. |
| This journal is © The Royal Society of Chemistry 2014 |