
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct core functionalisation of naphthalenediimides by iridium catalysed C–H borylation†
Catherine L.
Lyall
,
Camilla C.
Shotton
,
Monserrat
Pérez-Salvia
,
G.
Dan Pantoş
* and
Simon E.
Lewis
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: G.D.Pantos@bath.ac.uk; S.E.Lewis@bath.ac.uk
First published on 18th September 2014
Abstract
We report the first boron-substituted naphthalenediimides (NDIs), prepared by iridium catalysed C–H activation. When the NDI substrates bear N-benzyl substituents, the naphthyl NDI core is borylated in preference, suggestive of a directed borylation mechanism. Borylated NDIs are substrates for Suzuki–Miyaura couplings and borylation of an NDI bearing two inequivalent N-substituents has also been demonstrated.
Naphthalenediimides (NDIs) have been the focus of intense and increasing research activity in recent years. This is due to the very wide range of areas in which they have found application, including organic electronics,1 bulk heterojunction solar cells,2 artificial photosynthesis,3 synthetic pores and sensors,4 electrochemical DNA detection,5 DNA polyintercalation,6 G-quadruplex binding and antiproliferative agents,7 bioimaging,8 rotaxanes, catenanes and knots,9 dynamic systems and supramolecular assemblies,10 ligands for transition metals,11 metal–organic frameworks,12 organocatalysts13 and thermoresponsive materials.14 In each field it is desirable to access NDIs with specific substituents in order to modulate their properties and accordingly much synthetic effort is being directed towards this goal.
Synthesis of N-substituted NDIs is straightforward, as condensation of the corresponding primary amine with the commercially available 1,4,5,8-naphthalenetetracarboxylic dianhydride (1, NDA) reliably furnishes the desired NDIs; we have previously described the synthesis of non-symmetrically N-substituted NDIs 4 by two sequential condensations with two different amines in a one-pot reaction15 (Scheme 1).
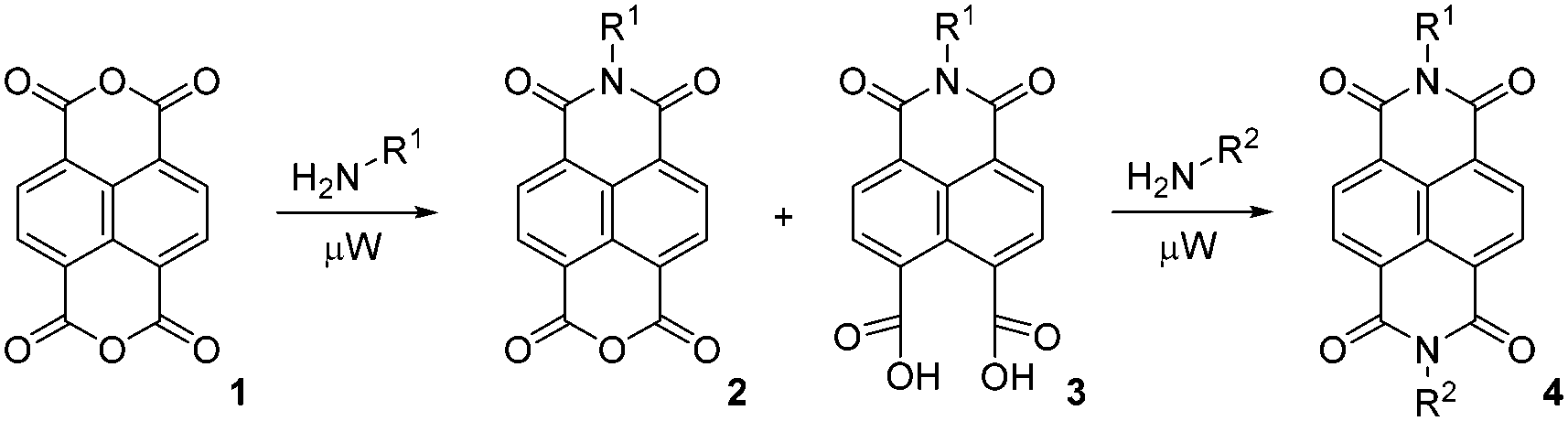 | ||
| Scheme 1 Synthesis of differentially N-substituted NDIs. | ||
In contrast, however, preparation of core-substituted NDIs (referred to in the literature as cNDIs) typically necessitates multistep procedures. Most reported routes to cNDIs involve preparing a core-functionalised NDA followed by the condensation with primary amines described above. For example, the first report of a cNDI, nearly 80 years ago,16 describes the preparation of 2,6-disubstituted NDIs from 2,6-dichloro-NDIs, prepared in turn from 2,6-dichloro-NDA. More recently, this route has been employed to access a diverse array of cNDIs.17 However, this approach is unavoidably hampered by the difficult access to 2,6-dichloro-NDA itself – this is prepared from pyrene in a four-step procedure that requires careful control of reaction temperature as well as the use of fuming HNO3/concentrated H2SO4 at 100 °C.17b More recently, direct bromination of NDA has been developed,17b with the resulting bromo-NDAs being further elaborated to cNDIs.18 This is a far more concise route to cNDIs, and can give access to monobromo, C2 symmetric dibromo and tetrabromo NDAs. However, it still necessitates the use of concentrated H2SO4 and/or oleum. The various routes to access cNDIs have recently been reviewed.19
A conceptually distinct approach to cNDIs is to functionalize directly the naphthyl core of a pre-formed core-unfunctionalized NDI. Examples of this approach are scant. To the best of our knowledge, there are five reports of nucleophilic or radical addition of amines to NDIs to afford amino- or heteroannellated NDIs;20 two further reports describe radical perfluoroalkylation of NDIs21 and Ru-catalysed C–H activation/styrene incorporation of NDIs22 respectively. We were interested in the direct introduction of a substituent on the naphthyl core which would allow for further elaboration to a wide selection of cNDIs. In this context, a cNDI bearing a boron-containing functional group was appealing, as the elaboration of these by Suzuki–Miyaura cross-coupling is well established (NDIs bearing core boryl substituents are unknown to date). Core-borylated NDIs would also be complementary to the known halo-NDIs/NDAs, insofar as the former would be nucleophilic partners for a cross-coupling, whereas the latter are electrophilic partners. C–H borylation catalysed by iridium has been extensively developed in recent years23 and this current paper describes our development of the C–H borylation of NDIs.
Encouraging precedent for our desired transformation was provided in the recent report of the Ir-catalysed C–H borylation of perylenediimides (PDIs, the higher rylene homologs of NDIs) with bis(pinacolato)diboron (B2pin2).24 The borylated PDIs indeed proved to be versatile synthetic intermediates,25 but the borylation reaction itself did however require forcing conditions, i.e. 48–96 h at 110 °C in 1,4-dioxane. In the first instance we adopted these same reaction conditions to attempt the borylation of NDIs. As test substrate we employed 5, bearing branched alkyl N-substituents to aid solubility (Scheme 2). Three discrete borylated species were identified: monoborylated NDI 6 (17% conversion) and isomeric diborylated NDIs 7 and 8 (76% combined conversion). Separation of 7 from 8 was not possible chromatographically. The structure of 6 was assigned on the basis of mass spectrometry and through the observation of four inequivalent carbonyl environments in the 13C-NMR spectrum. The structures of 7 and 8 were assigned on the basis of mass spectrometry and exhaustive NMR analysis of the mixture (see ESI†). Both C2-symmetric isomer 7 and CS-symmetric isomer 8 exhibit a single naphthyl resonance in the 1H-NMR spectrum. These are distinguishable and integrate in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (both before and after purification), implying that the second borylation event is entirely non-selective (ortho-diborylation being discounted on steric grounds). The 1H-NMR signal for the methylene group adjacent to nitrogen is also diagnostic: for 7 this is a doublet, but for 8, two doublets are observed as a result of the inequivalence of the two nitrogens in this molecule. It should be noted that the CS isomer 8 possesses a core substitution pattern that has only recently18a become accessible (indirectly) by the previously reported NDA bromination methodology.
:1 ratio (both before and after purification), implying that the second borylation event is entirely non-selective (ortho-diborylation being discounted on steric grounds). The 1H-NMR signal for the methylene group adjacent to nitrogen is also diagnostic: for 7 this is a doublet, but for 8, two doublets are observed as a result of the inequivalence of the two nitrogens in this molecule. It should be noted that the CS isomer 8 possesses a core substitution pattern that has only recently18a become accessible (indirectly) by the previously reported NDA bromination methodology.
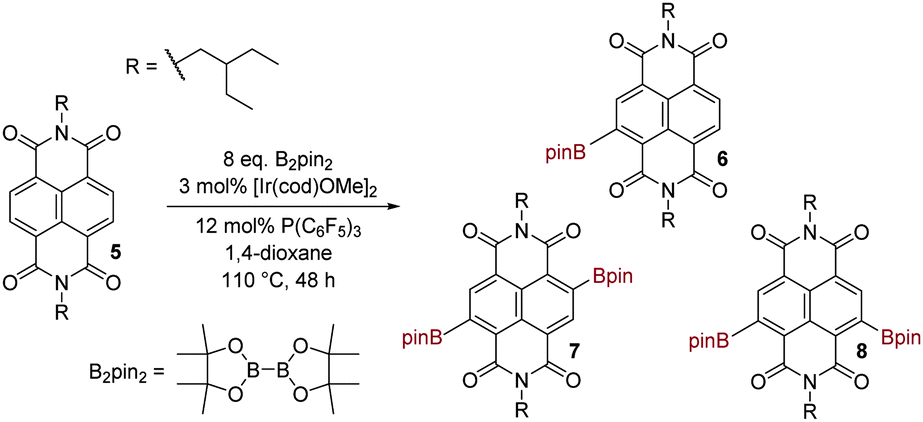 | ||
| Scheme 2 Iridium catalysed C–H borylation of an NDI under conditions of thermal heating. | ||
We next briefly explored the extent to which altering reaction time or stoichiometry affected the ratio of products 6:(7 + 8). As would be expected, a reduction in reaction time to 24 h increased the proportion of monoborylated product (28% conversion to 6) to diborylated products (64% conversion to 7 and 8, 1:1). A similar outcome was seen upon lowering the excess of B2pin2 to 4 equivalents, keeping reaction time at 48 h (50% conversion to 6 and 25% conversion to 7 and 8, 1:1).
We then considered the possibility of using an alternative solvent, since 1,4-dioxane is a group 2B carcinogen and is able to form peroxides. However, a replacement solvent had to meet several criteria: (a) have no aryl C–H bonds, to avoid competing borylation of the solvent; (b) have no ability to ligate the metal centre; (c) be capable of solvating the NDI substrates of notoriously poor solubility; (d) have a sufficiently high boiling point to allow reaction at elevated temperatures without dangerous pressure build-up; (e) be amenable to microwave heating and; (f) have an improved safety profile. Hexafluorobenzene was identified as meeting these criteria. Carrying out the reaction in Scheme 2, but with this solvent in place of dioxane led to a near-identical outcome: conversion to monoborylated 6 (18%) and diborylated 7 and 8 (76%, 1:1). Thus, all subsequent reactions were carried out in hexafluorobenzene.
In order to reduce the prohibitively long reaction times, we next employed microwave acceleration and elevated temperatures. Thus, the reaction depicted in Scheme 2, but using hexafluorobenzene as solvent at 140 °C for 2 h under microwave heating and with a smaller excess of boron source (4.0 eq. B2pin2), afforded borylated products in near quantitative overall conversion, i.e. monoborylated 6 (12% conversion) and diborylated 7 and 8 (85% conversion, 1:1). A screen of ligands commonly utilized for Ir-catalysed C–H borylation confirmed tris(pentafluorophenyl)phosphine to be optimal (Table 1).
| Ligand | Conversion to productsa,b | |
|---|---|---|
| Monoborylated NDI 6 (%) | Diborylated NDIs 7 & 8c (%) | |
|
a 4 eq. B2pin2, 3 mol% [Ir(cod)OMe]2, 12 mol% ligand, C6F6, 140 °C, 2 h, μW.
b Determined by 1H-NMR.
c (1:1) ratio.
d Yields in parentheses are for isolated products after chromatography.
|
||
| 2,2′-Bipyridine | 0 | 0 |
| 4,4′-Di-tert-butyl-2,2′-bipyridine | 6 | 0 |
| P(C6H5)3 | 44 | 35 |
| P(C6F5)3 | 12 (8)d | 85 (67)d |
We next probed the effects of additional changes to the excess of borylating agent used (Table 2); lowering this to 2.0 gave the greatest conversion to monoborylated 6. Mass balance was near quantitative and unreacted 5 could be recovered from the crude reaction mixture by selective crystallisation from acetonitrile prior to chromatography. The borylation methodology also proved applicable to other NDI substrates (Scheme 3).
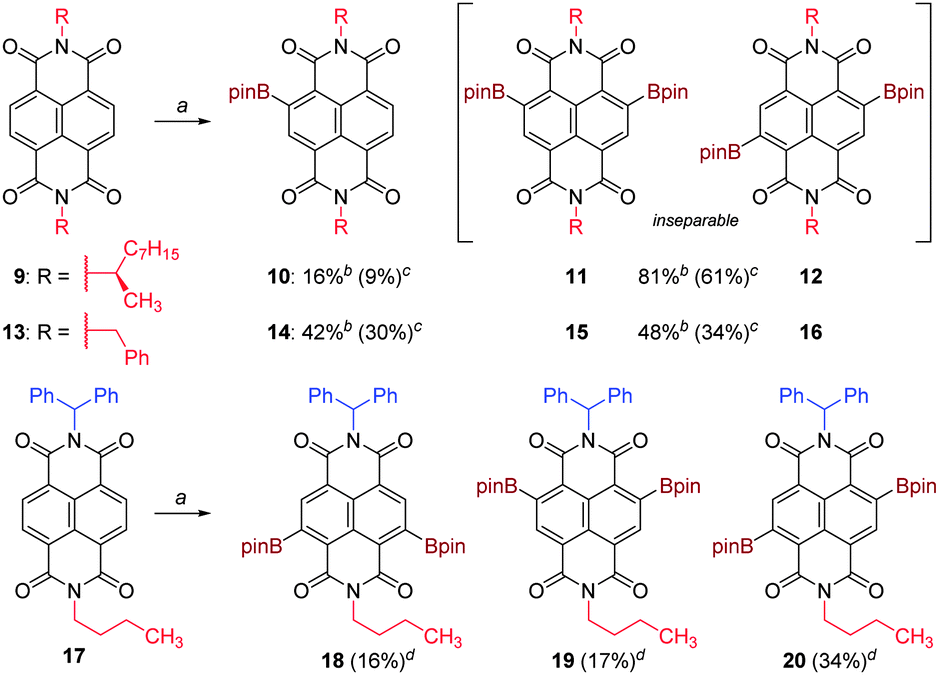 | ||
| Scheme 3 C–H Borylation of various NDI substrates. a4 eq. B2pin2, 3 mol% [Ir(cod)OMe]2, 12 mol% P(C6F5)3, C6F6, 140 °C, 2 h, μW. bConversion determined by 1H-NMR. cIsolated yield. dIsolated yield (purity ≥85%). | ||
Homochiral NDI substrate 9 underwent borylation with very similar conversions to 5, affording monoborylated 10 and diborylated 11 and 12 (inseparable, 1:1). Borylation of N,N′-dibenzyl NDI substrate 13 proceeded to a lesser extent under the same conditions, giving proportionately more monoborylated 14 to diborylated 15 and 16 (inseparable, 1:1). Notably, the borylation was entirely selective for the naphthyl core and no products arising from borylation of the benzyl groups were observed. This is strongly suggestive of the borylation being a directed process,26 which is precedented in the complete absence of bay-region functionalization in the reported borylation of PDIs.24 An NDI substrate bearing two inequivalent N-substituents, 17, was prepared as per Scheme 1, in order to probe the directing effects of these substituents. Upon subjecting 17 to the same reaction conditions, three diborylated products 18–20 were formed and isolated. In contrast to the preceding examples, no monoborylated products were isolated. The isomers 18:19:20 were formed in the statistical ratio 1:1:2, implying that despite the steric differences between the two N-substituents, both the first and second naphthyl borylation events were essentially nonselective. (Once again, no borylation of the aryl side-chain was observed). In contrast to the previous examples, isomers 18–20 proved sufficiently different in polarity that careful chromatography allowed for the isolation of each isomer in ≥85% purity, sufficient for their structures to be assigned unequivocally on the basis of 2D-NMR analysis (see ESI†).
To illustrate the synthetic utility of the borylated NDIs for accessing diverse chromophores, Suzuki–Miyaura cross-couplings were demonstrated with a variety of coupling partners. Monoborylated NDI 14 was used as a representative substrate (Fig. 1); a dual cross-coupling of 20 was also carried out to give 26. Both electron-rich and electron-poor aryl bromides are acceptable coupling partners, affording the isolated core-arylated NDIs in moderate to good yields. NDIs bearing a substituted phenyl group as their sole core substituent are an extremely scarce27 and an under-studied class of compounds. The absorption spectra for 21–26 are shown in Fig. 2. They all have absorption maxima between 330–390 nm which are essentially conserved, but 23 and 24 also have absorption maxima between 260–290 nm and methoxy-substituted 23 has a maximum at 459 nm, well into the visible region.
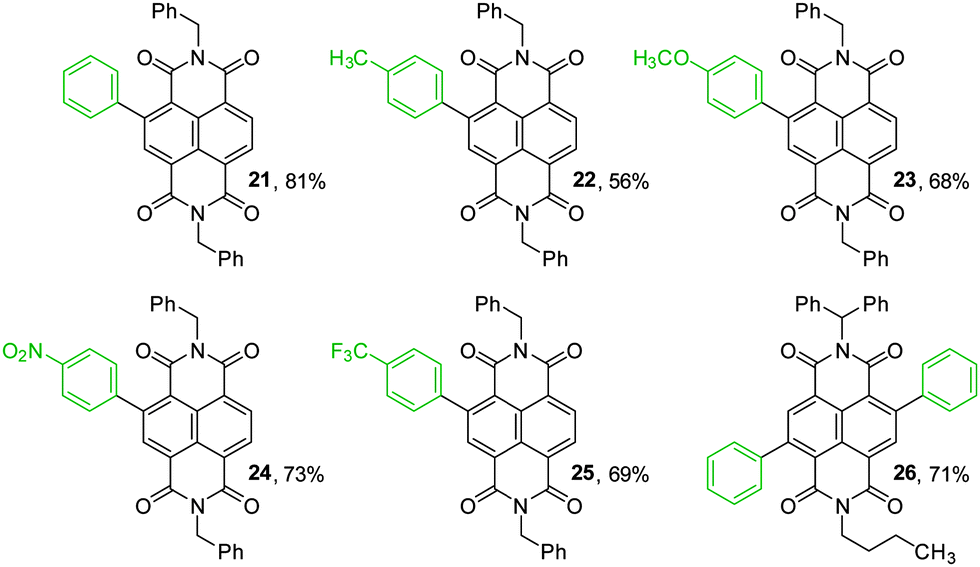 | ||
| Fig. 1 Products of Suzuki–Miyaura cross-coupling. Reaction conditions: 1.3 eq. Ar–Br per Bpin, 5.6 eq. K2CO3 per Bpin, 15 mol% Pd(PPh3)4, PhMe:H2O:EtOH 100:10:1, 80 °C, 16 h. Isolated yields given. | ||
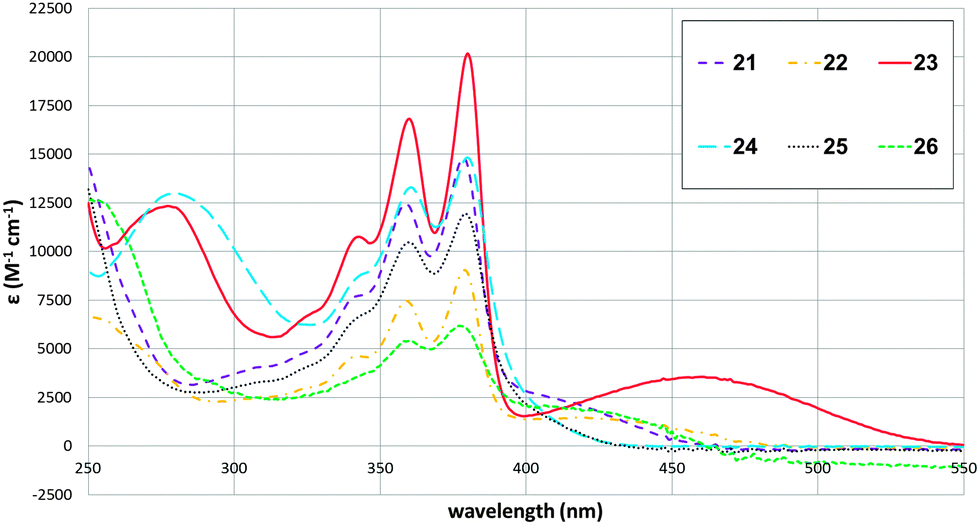 | ||
| Fig. 2 UV-Visible absorption spectra for 21–26 (solvent: CHCl3). | ||
In summary, we have demonstrated a concise and “core-selective” route to NDIs bearing boryl esters, which in turn are appropriate handles for further functionalisation. By these means, many previously unknown NDIs will be accessible. We anticipate this methodology will find multiple applications, given the plethora of uses for NDIs mentioned above.
Notes and references
- Review: X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960 CrossRef CAS PubMed.
- (a) A. Bolag, J. Lopez-Andarias, S. Lascano, S. Soleimanpour, C. Atienza, N. Sakai, N. Martin and S. Matile, Angew. Chem., Int. Ed., 2014, 53, 4890 CrossRef CAS PubMed; (b) N. Sakai, P. Charbonnaz, S. Ward and S. Matile, J. Am. Chem. Soc., 2014, 136, 5575 CrossRef CAS PubMed; (c) G. Sforazzini, E. Orentas, A. Bolag, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 12082 CrossRef CAS PubMed; (d) N. Sakai and S. Matile, Beilstein J. Org. Chem., 2012, 8, 897 CrossRef CAS PubMed; (e) S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84 CrossRef CAS PubMed.
- (a) A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791 CrossRef CAS PubMed; (b) N.-T. Lin, A. Vargas Jentzsch, L. Guenee, J.-M. Neudörfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121 RSC; (c) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed; (d) S. Hagihara, L. Gremaud, G. Bollot, J. Mareda and S. Matile, J. Am. Chem. Soc., 2008, 130, 4347 CrossRef CAS PubMed.
- (a) H. Takenaka, S. Sato and S. Takenaka, Electroanalysis, 2013, 25, 1827 CrossRef CAS; (b) S. Sato, K. Yamamura and S. Takenaka, Electroanalysis, 2013, 25, 1831 CrossRef CAS; (c) S. Sato and S. Takenaka, Anal. Sci., 2012, 28, 643 CrossRef CAS.
- (a) B. A. Ikkanda, S. A. Samuel and B. L. Iverson, J. Org. Chem., 2014, 79, 2029 CrossRef CAS PubMed; (b) A. Rhoden Smith and B. L. Iverson, J. Am. Chem. Soc., 2013, 135, 12783 CrossRef CAS PubMed; (c) A. Rhoden Smith, B. A. Ikkanda, G. G. Holman and B. L. Iverson, Biochemistry, 2012, 51, 4445 CrossRef CAS PubMed; (d) G. G. Holman, M. Zewail-Foote, A. R. Smith, K. A. Johnson and B. L. Iverson, Nat. Chem., 2011, 3, 875–881 CrossRef CAS PubMed.
- (a) I. Czerwinska, S. Sato, B. Juskowiak and S. Takenaka, Bioorg. Med. Chem., 2014, 22, 2593 CrossRef CAS PubMed; (b) S. Mpima, S. A. Ohnmacht, M. Barletta, J. Husby, L. C. Pett, M. Gunaratnam, S. T. Hilton and S. Neidle, Bioorg. Med. Chem., 2013, 21, 6162 CrossRef CAS PubMed; (c) F. Doria, M. Nadai, M. Folini, M. Scalabrin, L. Germani, G. Sattin, M. Mella, M. Palumbo, N. Zaffaroni, D. Fabris, M. Freccero and S. N. Richter, Chem. – Eur. J., 2013, 19, 78 CrossRef CAS PubMed; (d) M. Gunaratnam, S. Swank, S. M. Haider, K. Galesa, A. P. Reszka, M. Beltran, F. Cuenca, J. A. Fletcher and S. Neidle, J. Med. Chem., 2009, 52, 3774 CrossRef CAS PubMed.
- Z. Hu, G. D. Pantoş, N. Kuganathan, R. L. Arrowsmith, R. M. J. Jacobs, G. Kociok-Köhn, J. O'Byrne, K. Jurkschat, P. Burgos, R. M. Tyrrell, S. W. Botchway, J. K. M. Sanders and S. I. Pascu, Adv. Funct. Mater., 2012, 22, 503 CrossRef CAS.
- (a) N. Ponnuswamy, F. B. L. Cougnon, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2014, 136, 8243 CrossRef CAS PubMed; (b) A. J. Avestro, D. M. Gardner, N. A. Vermeulen, E. A. Wilson, S. T. Schneebeli, A. C. Whalley, M. E. Belowich, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 4442 CrossRef CAS PubMed; (c) H. Wilson, S. Byrne, N. Bampos and K. M. Mullen, Org. Biomol. Chem., 2013, 11, 2105 RSC; (d) N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783 CrossRef CAS PubMed; (e) H.-P. Jacquot de Rouville, J. Iehl, C. J. Bruns, P. L. McGrier, M. Frasconi, A. A. Sarjeant and J. F. Stoddart, Org. Lett., 2012, 14, 5188 CrossRef CAS PubMed; (f) S. K. Dey, A. Coskun, A. C. Fahrenbach, G. Barin, A. N. Basuray, A. Trabolsi, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2011, 2, 1046 RSC; (g) Y. Kasai, C. Sakamoto, N. Muroya, S.-i. Kato and Y. Nakamura, Tetrahedron Lett., 2011, 52, 623 CrossRef CAS PubMed.
- (a) S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. J. M. Frederix and R. V. Ulijn, Angew. Chem., Int. Ed., 2014, 53, 5882 CrossRef CAS PubMed; (b) K. Tambara, J.-C. Olsen, D. E. Hansen and G. D. Pantoş, Org. Biomol. Chem., 2014, 12, 607 RSC; (c) F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129 CrossRef CAS PubMed; (d) M. Kumar, N. Jonnalagadda and S. J. George, Chem. Commun., 2012, 48, 10948 RSC; (e) F. B. L. Cougnon, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443 CrossRef CAS PubMed; (f) N. Ponnuswamy, G. D. Pantoş, M. M. J. Smulders and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 566 CrossRef CAS PubMed; (g) A. R. Stefankiewicz, E. Tamanini, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2011, 50, 5725 CrossRef CAS PubMed; (h) T. W. Anderson, G. D. Pantoş and J. K. M. Sanders, Org. Biomol. Chem., 2011, 9, 7547 RSC; (i) E. Tamanini, N. Ponnuswamy, G. D. Pantoş and J. K. M. Sanders, Faraday Discuss., 2010, 145, 205 RSC; (j) H. Y. Au-Yeung, P. Pengo, G. D. Pantoş, S. Otto and J. K. M. Sanders, Chem. Commun., 2009, 419 RSC; (k) G. D. Pantoş, J.-L. Wietor and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 2238 CrossRef PubMed; (l) G. D. Pantoş, P. Pengo and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194 CrossRef PubMed ; erratum p. 2138.
- (a) F. N. Castellano, Dalton Trans., 2012, 8493 RSC; (b) F. Chaignon, M. Falkenström, S. Karlsson, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2007, 64 RSC.
- M. Pan, X.-M. Lin, G.-B. Li and C.-Y. Su, Coord. Chem. Rev., 2011, 255, 1921 CrossRef CAS PubMed.
- (a) Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101 CrossRef CAS PubMed; (b) H. Ke, L. Wang, Y. Chen, M.-J. Lin and J.-Z. Chen, J. Mol. Catal. A: Chem., 2014, 385, 26 CrossRef CAS PubMed.
- (a) C. Peebles, R. Piland and B. L. Iverson, Chem. – Eur. J., 2013, 19, 11598 CrossRef CAS PubMed; (b) K. R. Leight, B. E. Esarey, A. E. Murray and J. J. Reczek, Chem. Mater., 2012, 24, 3318 CrossRef CAS; (c) P. M. Alvey, J. J. Reczek, V. Lynch and B. L. Iverson, J. Org. Chem., 2010, 75, 7682 CrossRef CAS PubMed; (d) V. J. Bradford and B. L. Iverson, J. Am. Chem. Soc., 2008, 130, 1517 CrossRef CAS PubMed; (e) J. Q. Nguyen and B. L. Iverson, J. Am. Chem. Soc., 1999, 121, 2639 CrossRef CAS.
- (a) K. Tambara, N. Ponnuswamy, G. Hennrich and G. D. Pantoş, J. Org. Chem., 2011, 76, 3338 CrossRef CAS PubMed; (b) P. Pengo, G. D. Pantoş, S. Otto and J. K. M. Sanders, J. Org. Chem., 2006, 71, 7063 CrossRef CAS PubMed.
- (a) H. Vollmann, DE Pat.661756 1933; (b) H. Vollmann, H. Becker, M. Corell and H. Streeck, Liebigs Ann., 1937, 531, 1 CrossRef CAS.
- For selected examples, see: (a) A. Blaszczyk, M. Fischer, C. von Hänisch and M. Mayor, Helv. Chim. Acta, 2006, 89, 1986 CrossRef CAS; (b) C. Thalacker, C. Röger and F. Würthner, J. Org. Chem., 2006, 71, 8098 CrossRef CAS PubMed; (c) C. Thalacker, A. Miura, S. De Feyter, F. C. De Schryver and F. Würthner, Org. Biomol. Chem., 2005, 3, 414 RSC; (d) F. Würthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem. – Eur. J., 2002, 8, 4742 CrossRef.
- For selected examples, see: (a) R. Steyrleuthner, R. Di Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245 CrossRef CAS PubMed; (b) S.-L. Suraru, C. Burschka and F. Würthner, J. Org. Chem., 2014, 79, 128 CrossRef CAS PubMed; (c) Y. V. Suseela, M. Sasikumar and T. Govindaraju, Tetrahedron Lett., 2013, 54, 6314 CrossRef CAS PubMed ; corrigendum: Y. V. Suseela, M. Sasikumar and T. Govindaraju, Tetrahedron Lett., 2014, 55, 1959 Search PubMed; (d) M. Sasikumar, Y. V. Suseela and T. Govindaraju, Asian J. Org. Chem., 2013, 2, 779 CrossRef CAS; (e) K. Xiong and Y. Xiao, Tetrahedron Lett., 2013, 54, 3171 CrossRef CAS PubMed; (f) S.-L. Suraru and F. Würthner, J. Org. Chem., 2013, 78, 5227 CrossRef CAS PubMed; (g) J. Chang, Q. Ye, K.-W. Huang, J. Zhang, Z.-K. Chen, J. Wu and C. Chi, Org. Lett., 2012, 14, 2964 CrossRef CAS PubMed; (h) L. E. Polander, A. S. Romanov, S. Barlow, D. K. Hwang, B. Kippelen, T. V. Timofeeva and S. R. Marder, Org. Lett., 2012, 14, 918 CrossRef CAS PubMed; (i) W. Yue, J. Gao, Y. Li, W. Jiang, S. Di Motta, F. Negri and Z. Wang, J. Am. Chem. Soc., 2011, 133, 18054 CrossRef CAS PubMed; (j) T. D. M. Bell, S. Yap, C. H. Jani, S. V. Bhosale, J. Hofkens, F. C. De Schryver, S. J. Langford and K. P. Ghiggino, Chem. – Asian J., 2009, 4, 1542 CrossRef CAS PubMed; (k) R. S. K. Kishore, V. Ravikumar, G. Bernardinelli, N. Sakai and S. Matile, J. Org. Chem., 2008, 73, 738 CrossRef CAS PubMed; (l) C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070 CrossRef PubMed; (m) X. Gao, W. Qiu, X. Yang, Y. Liu, Y. Wang, H. Zhang, T. Qi, Y. Liu, K. Lu, C. Du, Z. Shuai, G. Yu and D. Zhu, Org. Lett., 2007, 9, 3917 CrossRef CAS PubMed.
- Reviews: (a) S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428 CrossRef CAS PubMed; (b) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC.
- (a) G. Rauch and S. Höger, Chem. Commun., 2014, 50, 5659 RSC; (b) H. Langhals, S. Christian and A. Hofer, J. Org. Chem., 2013, 78, 9883 CrossRef CAS PubMed; (c) C. Zhou, Y. Li, Y. Zhao, J. Zhang, W. Yang and Y. Li, Org. Lett., 2011, 13, 292 CrossRef CAS PubMed; (d) H. Langhals and S. Kinzel, J. Org. Chem., 2010, 75, 7781 CrossRef CAS PubMed; (e) H. Langhals and H. Jaschke, Chem. – Eur. J., 2006, 12, 2815 CrossRef CAS PubMed.
- Y. Li, C. Li, W. Yue, W. Jiang, R. Kopecek, J. Qu and Z. Wang, Org. Lett., 2010, 12, 2374 CrossRef CAS PubMed.
- J. E. Bullock, M. T. Vagnini, C. Ramanan, D. T. Co, T. M. Wilson, J. W. Dicke, T. J Marks and M. R. Wasielewski, J. Phys. Chem. B, 2010, 114, 1794 CrossRef CAS PubMed.
- Review: I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532 CrossRef CAS PubMed . A Ru-catalysed has also been reported: G. Battagliarin, C. Li, V. Enkelmann and K. Müllen, Org. Lett., 2011, 13, 3012 CrossRef PubMed.
- G. Battagliarin, Y. Zhao, C. Li and K. Müllen, Org. Lett., 2011, 13, 3399 CrossRef CAS PubMed.
- Review: A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC.
- (a) J. Gao, Y. Li and Z. Wang, Org. Lett., 2013, 15, 1366 CrossRef CAS PubMed; (b) K. Sekido, H. Nagasaka, M. Sekiya and K. Fukaya, Jp. Pat.2007148292; (c) I. A. Ronova, I. I. Ponomarev, A. Yu Kovalevsky and O. V. Shishkin, High Perform. Polym., 1999, 11, 355 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and spectra. See DOI: 10.1039/c4cc06522k |
| This journal is © The Royal Society of Chemistry 2014 |