
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand design for long-range magnetic order in metal–organic frameworks†
Davide
Tiana
,
Christopher H.
Hendon
and
Aron
Walsh
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.walsh@bath.ac.uk
First published on 23rd September 2014
Abstract
We report a class of ligands that are candidates to construct metal–organic frameworks with long-range magnetic order between transition metal centres. Direct quantum chemical calculations predict Néel temperatures exceeding 100 K for the case of Mn.
Metal–organic frameworks (MOFs) represent a diverse collection of materials featuring both inorganic and organic motifs, arranging in highly-ordered, and often porous arrays. Their chemical diversity and porosity has been subject to research efforts in catalysis,1–4 gas storage, separation and sensing,5–7 and recently in electrically conducting devices.8–11 Beyond these popular applications, MOFs also display interesting solid-state behaviour such as disorder,12 ion conduction,13 and magnetism.14,15
Magnetism in MOFs is particularly interesting as ordered arrays of magnetically active species pose opportunity for data storage, spin-frustrated catalysis and magnetosensing.16 Reports of magnetic MOFs are becoming more frequent;17–21 notable examples are that of HKUST-1 (featuring an antiferromagnetically coupled bis-Cu(II) paddlewheels, shown in Fig. 1b)22 and many Mn(II) based MOFs.23,24 There are also an increasing number of reports on tailor-made MOFs specifically designed to achieve ferroelectric, magnetic and multiferroic behaviour.25–27
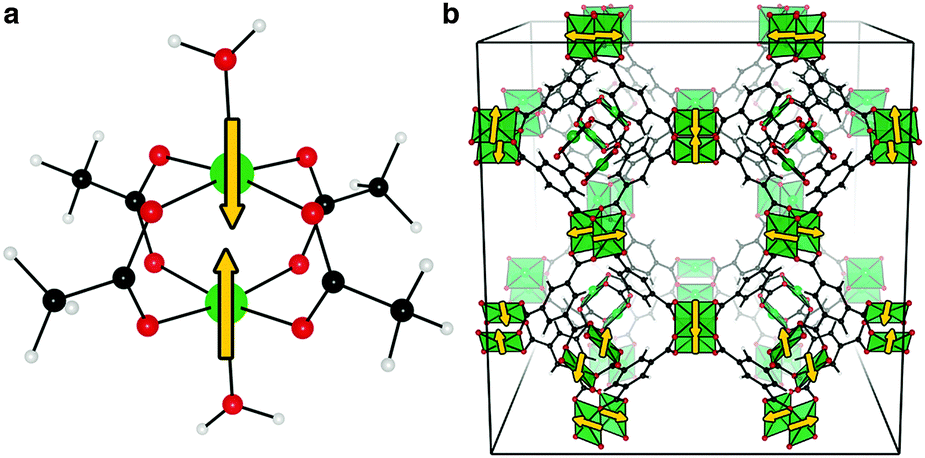 | ||
| Fig. 1 The Cu–Cu paddlewheel exhibits an antiferromagnetic (open-shell singlet) magnetic ground-state in both the single molecule cupric acetate dihydrate, a, and the periodic metal–organic framework, HKUST-1, b. Schematic spin alignments are shown with arrows. | ||
The development of ligands to exploit magnetism in hybrid materials is an active area.28,29 For example, magnetic frameworks have been obtained using N-donor heterocycles30 with polycarboxylates.31,32 Despite this progress, critical temperatures have been very low to date. While the understanding of magnetism in inorganic materials is well developed, for hybrid materials there are no design principles for obtaining high Curie (TC) or Néel (TN) temperatures.33
In general, magnetism in materials can be described by the coupling of individual magnetic moments through an exchange interaction (J). A simple example is shown in Fig. 1a (and Fig. 2), a dimer with two unpaired electrons can have two spin configurations, where J = (ES − ET)/2 is the energy difference between the singlet (S) and the triplet (T) states. If J > 0 the parallel spin configuration (ferromagnetic state, FM) is favoured. Conversely, the anti-parallel (antiferromagnetic state, AFM) spin configuration is favoured if J < 0. For example, cupric acetate dihydrate (Fig. 1a), a molecular analogue of the HKUST-1 hybrid framework, is an organometallic molecule with a range of accessible spin configurations.34,35
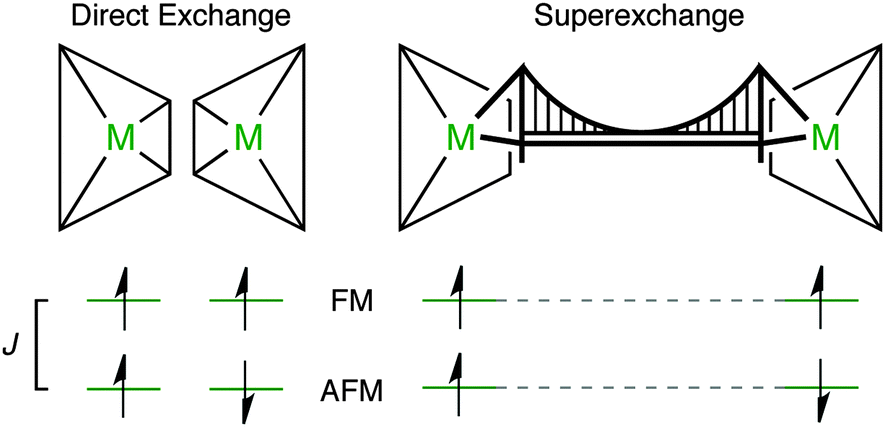 | ||
| Fig. 2 The exchange energy, J, is determined by the energy difference between the singlet and the triplet states for a spin 1/2 dimer. Direct exchange is observed in spin-polarised metals in close proximity (e.g. the paddlewheel motif found in some metal–organic frameworks). Super-exchange is mediated by a diamagnetic bridging motif (e.g. the oxide ion in Mn–O–Mn). | ||
Direct magnetic exchange only occurs when there is electronic wavefunction overlap between the two magnetic sites. In most MOFs, the metal–metal distance will be too large for this to occur. In contrast, super-exchange can be active over longer distances.36 Usually, metal cations are coupled through a diamagnetic anion (which we have schematically represented as a bridging motif, Fig. 2). A typical example is MnO which displays AFM order with TN = 118 K.37 Each Mn(II) centre has a formal electronic configuration of 3d5 and the Mn–O–Mn angle is 180° in the rocksalt structure, which are both optimal for super-exchange following the Goodenough–Kanamouri rules.38,39 Another criterion is orbital symmetry: here, the Mn eg ligand field combination effectively overlaps with the oxide 2p orbitals. The aim of this study is to identify an organic ligand suitable for mediating long-range super-exchange in metal–organic frameworks.
In the solid state, the exchange energy must be generalised to include factors such as the spin quantum number (S), the coordination number (z) and the site occupation (N):
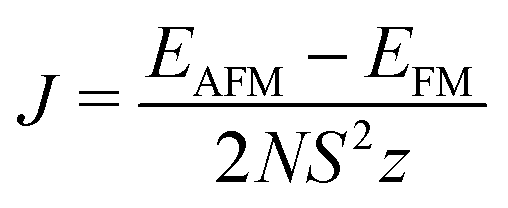
This exchange energy can then be used in the standard Heisenberg Hamiltonian, from which the critical temperature is recovered within the mean field approximation (Van Vleck's formula):40
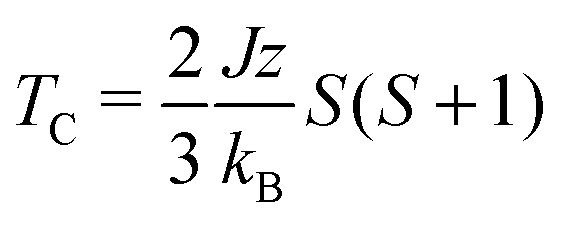
In the HKUST-1 framework, a strong direct exchange interaction within a Cu(II)–Cu(II) motif is present, but the cupric pairs are too far apart (>8 Å) to display any long-range magnetic order. Indeed, super-exchange with carboxylates tends to occur only within the same COO head.15 For the direct interactions between the two 3d9 centres, an exchange constant J = −10 meV is calculated with an associated critical temperature of 348 K. This critical temperature is consistent with the molecular analogue copper acetate, where TN ∼ 280 K.
For effective super-exchange, as discussed earlier, the valence shell of the transition metal (TM) should be half-filled (e.g. the Mn(II) or Fe(III) ions), and the TM–ligand–TM angle should be close to 180°.38,39 Mn(II) is therefore an ideal metal, which readily forms metal–organic frameworks and can adopt tetrahedral or octahedral coordination environments. In MnO, the 2p atomic orbitals of a single oxide ion facilitate super-exchange, while in a metal–organic framework, the coupling must be along the molecular orbitals of the entire ligand.
An ideal ligand would have an extended π system and feature orthogonal terminal groups. The ligands most commonly used in MOFs are either planar or have frontier orbitals localised on a central aromatic ring. However, such characteristics are embodied in oligoynes terminated in carboxylate groups, which are conjugated molecules with a helical continuous π wavefunction.44 Their structural linearity may also allow so-called electron ‘hopping’ between metal centres. To our knowledge, there is only one example of a MOF featuring this ligand; however, it was constructed with the diamagnetic Zn(II) ion.45
Based on the considerations presented above, we have constructed a series of model systems combining Mn(II) and oligoyne dicarboxylates (Fig. 3). These one-dimensional coordination polymers are used as a model of the typical connectivity found in MOFs. In the smallest system, the Mn centres are linked by acetylene dicarboxylate (ACDC-1), which features orthogonal carboxylate motifs, and a single alkyne. The system is expanded by increasing the concatenation of the alkyne (i.e. the di-yne and tri-yne are reported as ACDC-2 and ACDC-3, respectively).
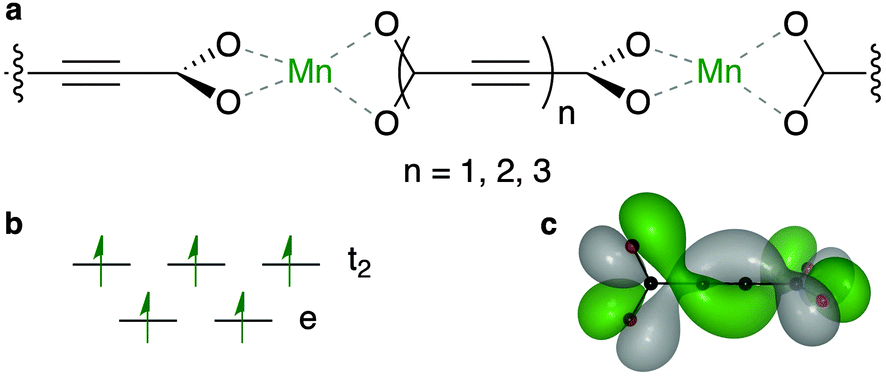 | ||
| Fig. 3 Schematic representation of the Mn–ACDC–Mn conformation, a, that satisfies the basic requirements for long-range super-exchange. The smallest system (d(M–M) = 8.8 Å) is composed of high-spin Mn(II) ions in a tetrahedral ligand field, b, and one of the degenerate highest occupied molecular orbitals of the helically conjugated ACDC-1, c (isovalue 1 × 10−2 e A−3). The combination of the two motifs to form a 1D framework results in a predicted Néel temperature of 117 K comparable to that of MnO. | ||
Analysis of the electronic structure reveals that five highest occupied crystal orbitals, are produced by the tetrahedral crystal field splitting of the Mn d shell. This TM orbital overlap with the O pz produces a bonding interaction (see the ESI† for plots of all frontier orbitals). The lowest unoccupied crystal orbital is ligand centred, featuring the helical topology determined by single molecule calculations of the free ligand.44 The higher energy unoccupied states are centred on the Mn–O interaction.
Due to the coordination environment, only orbitals with t2 symmetry can yield electron hopping from one metal center to the other via O p orbitals (i.e. providing super-exchange interactions). Moreover, it has been recently emphasised how the correct directionality is also required for crystalline orbital to be fully delocalized through a coordination polymer.46 Only the Mn dxz and dyz orbitals are active in our model system. These orbitals can combine with the helical highest molecular orbital of ACDC (Fig. 3c). The associated spin density (Fig. 4) illustrates the strong polarisation of the terminal carboxylate groups of the ligand. The changes in magnetic structure are described in Table 1; the electronic structure changes are further explored in the ESI.†
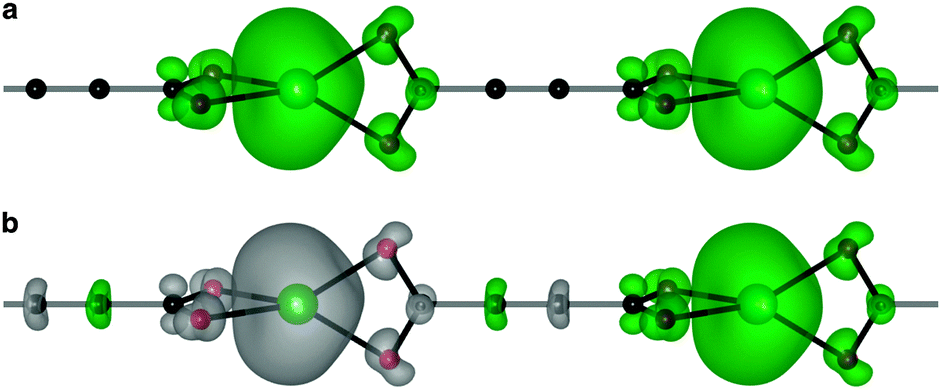 | ||
| Fig. 4 The ferromagnetic (a) and antiferromagnetic (b) spin densities (ρ↑–ρ↓) of MnACDC-1 (isovalue 3 × 10−2 e Å−3). The ground-state magnetic configuration is AFM; note the strong polarisation of the terminal carboxylate groups and the spin contributions from the diamagnetic ligand for the AFM case. | ||
| n | d(M–M) | ΔE | J | T N |
|---|---|---|---|---|
| 1 | 8.8 | −22 | −0.86 | 117 |
| 2 | 11.4 | −10 | −0.38 | 51 |
| 3 | 14.0 | −5 | −0.20 | 26 |
As a result of the half-filled d shell of Mn, and the Pauli exclusion principle, the FM configuration is disfavoured. In the most extreme case, MnACDC-1 (Fig. 4), ΔE = −22 meV, with the AFM state being favoured (i.e. ΔE = EAFM − EFM). The strength of the interaction is remarkable given the large metal–metal separation. The associated exchange constant is −0.86 meV, which corresponds to a mean field TN of 117 K (Table 1) for this model system. This value is remarkably similar to the inorganic solid MnO, where the Mn–Mn separation is close to 3 Å.
The behaviour predicted for MnACDC-1 holds true for longer oligoynes, with J decreasing proportional to d(M–M). The exponential decay is characteristic of a super-exchange interaction47 (see ESI†). Examining MnACDC-2, d(M–M) increases to 11.4 Å. Similar to the monoalkyne, the same local magnetic moment is calculated for Mn(II) and the AFM state is the most stable; ΔE = −10 meV and TN = 51 K. From the electronic point of view, the main difference between ACDC-1 and 2 is that the occupied orbitals are destabilised due to the longer π system. There is an associated reordering of the crystalline orbitals, with the ligand states occupying the higher energy frontier orbitals (see ESI†). This change shields the two metals, reducing their magnetic coupling. The effect is further enhanced for ACDC-3 where d(M–M) increases to 14 Å. Here the energy difference reduces to 5 meV, corresponding to TN = 27 K.
To demonstrate the generality of the approach, additional calculations were performed on an analogous (n = 1) Mn polymer with octahedral coordination (via two axial water molecules). Here the energy difference is 19 meV, corresponding to TN = 105 K. Full details are given in the ESI.†
In summary, by considering the type of magnetic interactions that can take place in metal–organic frameworks, a series of simple rules for facilitating long-range magnetic order have been presented. We have shown that the paddlewheel motif, found in MOFs such as HKUST-1, can exhibit a strong direct exchange interaction that may later be harnessed for magnetosensing. Further, the principles of super-exchange have been used to tailor hybrid materials with high critical temperatures, comparable to magnetic oxides, even though the separation between the spin polarised metals is in excess of 8 Å. It is evident that combining extended conjugated linear organic molecules with transitions metals is an effective approach to achieve long-range hybrid super-exchange. We hope that this work will stimulate experimental efforts to synthesise three-dimensional frameworks based on these components.
Computational details. All calculations were performed within the solid-state quantum chemical code Quantum Espresso (version 5.0.2)48 at the density functional level of theory. The semi-local PBEsol exchange correlation functional was employed for ionic relaxation,49 using norm-conserving pseudopotentials with a cut-off of 80 Ry. Dispersion force corrections were also employed.50,51
Nine k-points along the polymer axis were generated with the Monkhorst–Pack grid.52 The force and energy thresholds were set to 5 × 10−3 eV and 5 × 10−4 eV, respectively, for geometry optimisation. The spin state of single atoms were allowed to relax during the optimisation with an SCF convergence threshold of 1 × 10−6 eV.
In order to test the dependence on the treatment of electron exchange and correlation, additional calculations were performed using the local LDA53 and non-local hybrid HSE06 level of theory.54 We also tested the inclusion of spin–orbit coupling, which made no notable difference to the results. A further comparison with PAW pseudopotential was also performed (see the ESI† for further details).
This study was inspired by A. K. Cheetham's lecture at MC11. C.H.H. thanks N. F. Chilton for useful discussions on magnetism. D.T. and C.H.H. are funded by the ERC (Grant No. 277757). A.W. acknowledges support from the Royal Society University Research Fellowship scheme. The work benefits from the high performance computing facility at the University of Bath. Access to the HECToR supercomputer was facilitated through membership of the HPC Materials Chemistry Consortium (EP/F067496). Images of chemical structures and orbitals were made using the VESTA software.55 This work made use of the software package GNU Octave, and the authors are grateful for the support of the Octave development community.56
References
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- K. T. Butler, C. H. Hendon and A. Walsh, J. Am. Chem. Soc., 2014, 136, 2703 CrossRef CAS PubMed.
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513 RSC.
- D. Britt, D. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623 CrossRef CAS PubMed.
- J. B. Decoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695 CrossRef CAS PubMed.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem. – Eur. J., 2011, 17, 11372 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120 RSC.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS PubMed.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994 RSC.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913 RSC.
- D.-F. Weng, Z.-M. Wang and S. Gao, Chem. Soc. Rev., 2011, 40, 3157 RSC.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC.
- Given that electrically conducting MOFs are rare, there is an appeal to making a device based on change in magnetism rather than voltage.
- L. M. Toma, C. Ruiz-Pérez, J. Pasán, W. Wernsdorfer, F. Lloret and M. Julve, J. Am. Chem. Soc., 2012, 134, 15265 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef.
- Z. Wang, P. Jain, K. Choi and J. V. Tol, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 224406 CrossRef.
- Y. Tian, W. Wang, Y. Chai, J. Cong, S. Shen, L. Yan, S. Wang, X. Han and Y. Sun, Phys. Rev. Lett., 2014, 112, 017202 CrossRef.
- P. J. Saines, P. T. Barton, M. Jura, K. S. Knight and A. K. Cheetham, Mater. Horiz., 2014, 1, 332–337 RSC.
- S. S. Chui, S. M. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625 CrossRef CAS PubMed.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dincǎ, J. Am. Chem. Soc., 2014, 136, 3334 CrossRef CAS PubMed.
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163 CrossRef CAS PubMed.
- K.-L. Hu, M. Kurmoo, Z. Wang and S. Gao, Chem. – Eur. J., 2009, 15, 12050 CrossRef CAS PubMed.
- A. Stroppa, P. Jain, P. Barone, M. Marsman, J. M. Perez-Mato, A. K. Cheetham, H. W. Kroto and S. Picozzi, Angew. Chem., Int. Ed., 2011, 50, 5847 CrossRef CAS PubMed.
- X.-Y. Wang, Z.-M. Wang and S. Gao, Chem. Commun., 2008, 281–294 RSC.
- M. Pilkington, M. Gross, P. Franz, M. Biner, S. Decurtins, H. Stoeckli-Evans and A. Neels, J. Solid State Chem., 2001, 159, 262 CrossRef CAS.
- K. S. Murray, Eur. J. Inorg. Chem., 2008, 3101 CrossRef CAS.
- M. Kurmoo, H. Kumagai, S. M. Hughes and C. J. Kepert, Inorg. Chem., 2003, 42, 6709–6722 CrossRef CAS PubMed.
- N. Guillou, C. Livage and G. Frey, Eur. J. Inorg. Chem., 2006, 4963–4978 CrossRef CAS.
- A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58 CrossRef CAS PubMed.
- E. I. Solomon, B. L. Hemming and D. E. Root, Electronic Structures of Active Sites in Copper Proteins: Coupled Binuclear and Trinuclear Cluster Sites, Springer Netherlands, 1993, pp. 3–5 Search PubMed.
- P. K. Ross, M. D. Allendorf and E. I. Solomon, J. Am. Chem. Soc., 1989, 111, 4009 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1950, 79, 350 CrossRef.
- J. B. Goodenough, Magnetism and the Chemical Bond, Wiley, Cambridge, MA, USA, 1963 Search PubMed.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117 CrossRef CAS PubMed.
- S. Blundell, Magnetism in Condensed Matter, Oxford University Press, Oxford, OX2 6DP UK, 2001 Search PubMed.
- P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975, 97, 4884 CrossRef CAS.
- H.-J. Koo, M.-H. Whangbo, P. D. VerNooy, C. C. Torardi and W. J. Marshall, Inorg. Chem., 2002, 41, 4664 CrossRef CAS PubMed.
- H.-J. Koo and M.-H. Whangbo, Inorg. Chem., 2000, 39, 3599 CrossRef CAS.
- C. H. Hendon, D. Tiana, A. T. Murray, D. R. Carbery and A. Walsh, Chem. Sci., 2013, 4, 4278 RSC.
- D. Tranchemontagne, J. Hunt and O. Yaghi, Tetrahedron, 2008, 64, 8553 CrossRef CAS PubMed.
- D. Tiana, C. H. Hendon, A. Walsh and T. P. Vaid, Phys. Chem. Chem. Phys., 2014 10.1039/C4CP00008K.
- R. E. Coffman and G. R. Buettner, J. Phys. Chem., 1979, 83, 2387 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- J. W. Eaton, D. Bateman and S. Hauberg, GNU Octave version 3.0.1 manual: a high-level interactive language for numerical computations, CreateSpace Independent Publishing Platform, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc06433j |
| This journal is © The Royal Society of Chemistry 2014 |