
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented formation of polycyclic diazadiborepine derivatives through cage deboronation of m-carborane†‡
Nicole
Harmgarth
,
Cristian G.
Hrib
,
Volker
Lorenz
,
Liane
Hilfert
and
Frank T.
Edelmann
*
Chemisches Institut der Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, D-39106 Magdeburg, Germany. E-mail: frank.edelmann@ovgu.de; Fax: +49-391-6712933
First published on 10th September 2014
Abstract
An unprecedented deboronation reaction of icosahedral carboranes is described, in which a BH group of m-carborane is detached from the cage and incorporated into an unusual nido-carborane-anellated diazadiborepine ring system.
Icosahedral carboranes form an exciting and highly important class of inorganic cage compounds due to their numerous important applications, including energy storage, optoelectronics, nanomaterials, radiopharmaceuticals, medicine, and boron neutron capture therapy.1,2 Deboronation, i.e. the removal of a vertex from a carborane cage under formation of an anionic nido-C2B9 cluster, is one of the most important and longest known reactions of icosahedral carboranes.3,4 Deboronation of closo-carboranes is normally achieved by treatment with strong Lewis bases (neutral or anionic) such as fluorides,5 alkoxides,3b amines,6 or N-heterocyclic carbenes.7 Deboronation rates have been reported to decrease from o- to p-carborane.5b According to the generally accepted mechanism (Scheme 1, illustrated for o-carborane (1)), deboronation starts with nucleophilic attack at one of the most positive boron vertices, which is followed by attack of a second nucleophile at the same B atom.4
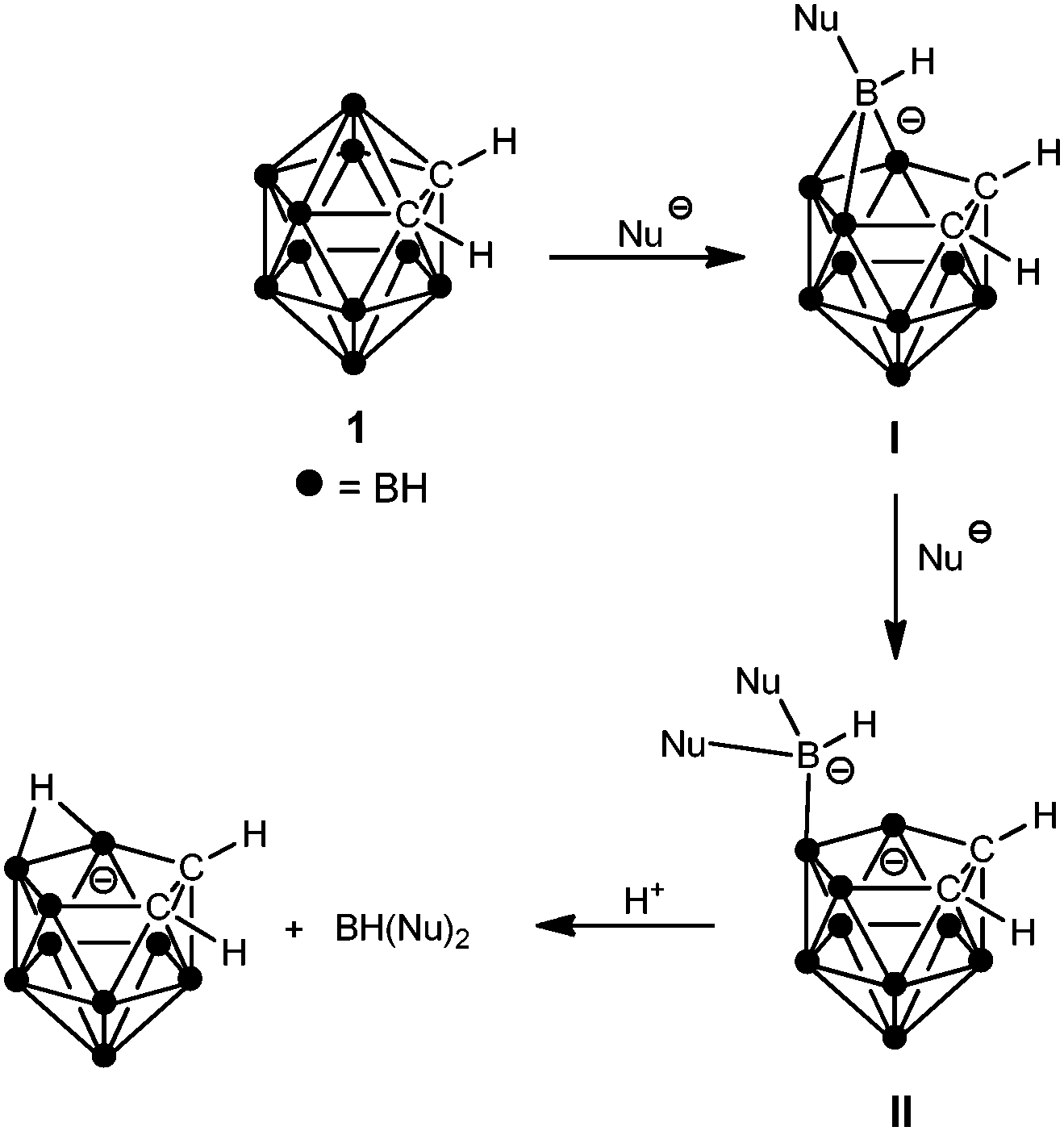 | ||
| Scheme 1 Mechanism of the deboronation of o-carborane (1) with anionic nucleophiles (taken from ref. 4). | ||
Protonation of the resulting negatively charged nido-cluster leads to elimination of BH(Nu)2 which can undergo further reactions with nucleophiles.7,8 For example, in fluoride ion-initialized deboronation reactions, the fluoroborate anion HOBHF2− has been detected as monoborane by-product.9 On rare occasions, it has been possible to isolate type I (e.g. Nu = (Me2N)3PNH,8a,10 pyridine,8b N-heterocyclic carbene7) intermediates, thereby backing the proposed mechanism experimentally. In all these intermediates, the BH unit bearing the nucleophile(s) remains attached to the original vertex of the carborane cage.
Recently, carboranylamidinates, a novel type of o-carborane-based chelating ligands, have been discovered in our lab.11 Carboranylamidinate anions combine the versatile characteristics of carboranes and the widely employed amidinate anions12 into one system. The general synthetic route to lithium carboranylamidinates involves in situ-preparation of monolithio-o-carborane, followed by treatment with a N,N′-diorganocarbodiimide, R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–R (R = iPr, cyclohexyl (Cy)) (Scheme 2). Quite unique and typical for the new carboranylamidinates is the C,N-chelating coordination mode while retaining an NH-functionality.11
CN–R (R = iPr, cyclohexyl (Cy)) (Scheme 2). Quite unique and typical for the new carboranylamidinates is the C,N-chelating coordination mode while retaining an NH-functionality.11
 | ||
| Scheme 2 Synthesis of lithium-o-carboranylamidinates from o-carborane (1). | ||
Following our initial report, the chemistry of carboranylamidinates has been rapidly extended to various main group element derivatives as well as early and late transition metal complexes, some of them showing promising catalytic activities.13 It was also demonstrated that the carboranylamidinate system can be modified by introducing thiolate or selenolate functional groups or by designing novel nido-carboranylamidinate ligands.14 Thus far, the synthetic protocol leading to carboranylamidinates (Scheme 2) has always been limited to the use of o-carborane (1) as starting material. This led us to the question of whether a similar chemistry could be developed around m-carborane (3). Here we report about the highly surprising and unexpected outcome of initial experiments in that direction.
m-Carborane (3) was mono-metalated in the usual manner11 using 1 equiv. of n-butyllithium followed by treatment of the intermediate lithio-m-carborane with 1 equiv. of N,N′-diisopropylcarbodiimide. Crystallization from either THF or diethyl ether afforded large, colorless single crystals in estimated yields around 30–40%. However, under these conditions, fairly large amounts of unreacted m-carborane could be detected in the reaction mixtures. Initially, analytical and spectroscopic data did not allow unambiguous elucidation of the isolated products. Fortunately, in both cases well-formed single crystals were readily available. X-ray diffraction studies revealed the presence of the unexpected products 4a and 4b as illustrated in Scheme 3. Accordingly, the outcome of these reactions can be described as follows: Unlike the formation of carboranylamidinates from o-carborane (1) according to Scheme 2, in situ-prepared monolithio-m-carborane reacts with 2 equiv. of N,N′-diisopropylcarbodiimide. This accounts for the presence of unreacted m-carborane in the original 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction mixture. Instead of a simple addition of the carborane anion to the central carbodiimide carbon alone, subsequent deboronation occurs and a BH unit is detached from the carborane cage. The following step is unprecedented in the chemistry of icosahedral carboranes, in that the BH unit is not eliminated as a monoborane by-product but retained in a newly formed seven-membered diazadiborepine ring system which is assembled in the periphery of the resulting nido-carborane cage through coupling of two carbodiimides. The result is the formation of an unusual nido-carborane-anellated diazadiborepine ring system which is isolated in the form of its lithium salt solvated with either THF (4a) or diethylether (4b) after recrystallization from the respective solvent (Scheme 3).
:1 reaction mixture. Instead of a simple addition of the carborane anion to the central carbodiimide carbon alone, subsequent deboronation occurs and a BH unit is detached from the carborane cage. The following step is unprecedented in the chemistry of icosahedral carboranes, in that the BH unit is not eliminated as a monoborane by-product but retained in a newly formed seven-membered diazadiborepine ring system which is assembled in the periphery of the resulting nido-carborane cage through coupling of two carbodiimides. The result is the formation of an unusual nido-carborane-anellated diazadiborepine ring system which is isolated in the form of its lithium salt solvated with either THF (4a) or diethylether (4b) after recrystallization from the respective solvent (Scheme 3).
 | ||
| Scheme 3 Formation of the polycyclic diazadiborepines 4a and 4b from m-carborane. | ||
Fig. 1 displays the molecular structure of the THF-adduct 4a. The molecular structure of 4b differs only in the coordinated solvent at Li (see ESI‡ for full structural details of both 4a and 4b). The newly formed seven-membered diazadiborepine ring (C1–C3–N2–B11–C10–B6) is anellated to a B–C bond of the nido-carborane cage. With C3–N1 1.2891(19) Å and C10–N4 1.2773(19) Å the two exocyclic CN bonds clearly have double bond character, whereas the bonds within the ring are single bonds (C3–N2 1.4106(19), C10–N3 1.427(2) Å). Lithium interacts with B5 (2.710(3) Å) and N1 (1.999(3) Å), thereby forming a five-membered C2BNLi chelate ring. Two THF ligands complete the distorted tetrahedral coordination geometry around Li. After the crystal structures of 4a and 4b had been unequivocally established, it was shown that by using the proper 1:2 stoichiometry, the reaction reproducibly provided 4a and 4b in moderate (4b: 51%) to good (4a: 67%) isolated yields. Moreover, the compounds were thoroughly characterized by spectroscopic and analytical methods (see ESI‡).
 | ||
| Fig. 1 Molecular structure of 4a. Selected bond lengths [Å] and angles [°]: N1–C3 1.2891(19), N1–Li 1.999(3), N2–C3 1.4106(19), N2–B11 1.437(2), N3–B11 1.418(2), N3–C10 1.427(2), N4–C10 1.2773(19), C1–C3 1.491(2), C10–B6 1.673(2), B5–Li 2.710(3); N1–C3–N2 126.41(14), N1–C3–C1 120.42(14), N2–C3–C1 113.02(12), N3–B11–N2 130.11(15). | ||
In a subsequent experiment it was shown that the analogous reaction with N,N′-dicyclohexylcarbodiimide proceeds in the same manner (Scheme 4). In this case, X-ray quality single crystals could be obtained from DME (=1,2-dimethoxyethane). Full spectroscopic characterization of the DME adduct 5 already indicated a similar outcome of this reaction as with iPr–NCN–iPr.
 | ||
| Scheme 4 Formation of the polycyclic diazadiborepine 5 from m-carborane. | ||
Once again, deboronation and carbodiimide coupling under formation of a diazadiborepine had occurred, underlining the fact that this novel reaction is quite general in nature. The NMR spectra of 5 (1H, 7Li, 11B, 13C) showed far-reaching similarity with those of 4a and 4b (apart from the different substituents). For example, in the 1H NMR spectrum of 5, the BH unit in the seven-membered ring gives rise to a broad singlet at δ 4.03 ppm. However, a single-crystal X-ray diffraction study of 5 revealed interesting structural differences (Fig. 2 and ESI‡). One of these differences is that Li interacts with two cage boron atoms (Li–B5 2.663(3), Li–B6 2.637(3) Å). Moreover, the electron distribution within one of the amidinate moieties (N3–C4–N4) differs significantly from that in 4a and 4b. Whereas the diazadiborepine ring in 4a and 4b comprises two exocyclic CN double bonds (vide supra), there is a bond between N4 and B10 in 5 which leads to a seven-membered open face of the nido-carborane cluster. As a consequence, bond delocalization in the amidinate unit is observed which results in equilibrated C–N distances (C4–N3 1.3400(16) and C4–N4 1.3178(16) Å). Whether or not these structural differences are a consequence of the different steric demand of the substituents (isopropyl vs. cyclohexyl) remains speculative at this stage.
 | ||
| Fig. 2 Molecular structure of 5. Selected bond lengths [Å] and angles [°]: B3–C4 1.5438(18), B5–Li1 2.663(3), B6–Li1 2.637(3), B10–N4 1.5440(17), B11–N2 1.4756(17), B11–N3 1.5438(17), Li1–N1 2.011(2), N1–C3 1.2894(16), N2–C3 1.4063(15), N4–C4 1.3178(16), C4–N3 1.3400(16), C1–C3 1.4989(16); C4–B3–C1 104.68(10), N2–B11–N3 118.36(11), N4–C4–N3 137.04(12), C4–N3–B11 106.11(10), C3–N2–B11 118.46(10). | ||
Based on the established knowledge about carboranylamidinate formation11 and deboronation reactions4 it appears reasonable to propose the mechanism illustrated in Scheme 5 for the formation of the novel diazadiborepines 4 and 5. Monolithiation of m-carborane (3) with 1 equiv. of n-butyllithium affords intermediate A which will add to the carbodiimide carbon to afford carboranylamidinate B. B–N bond formation could then activate the BH unit in the hydridoborate intermediate C for deboronation and diazadiborepine formation initialized by the second equivalent of carbodiimide.
 | ||
| Scheme 5 Proposed mechanism for the formation of the polycyclic diazadiborepines 4a, 4b, and 5 from m-carborane. | ||
In summarizing the results reported here, we have discovered an unprecedented deboronation reaction of icosahedral carboranes in which a BH group is detached from the cage and incorporated into a nido-carborane-anellated diazadiborepine ring. The reactions illustrated in Schemes 3 and 4 clearly open up a new field of carborane chemistry. Future work will show if and how the readily accessible lithium salts 4 and 5 can be employed as starting materials for novel carborane-based polycycles incorporating other main group or transition metals.
Financial support by the Otto-von-Guericke-Universität Magdeburg is gratefully acknowledged. We also thank Professor Edgar Haak for valuable discussions.
Notes and references
- M. F. Hawthorne, Advances in Boron Chemistry, Special Publication No. 201, Royal Society of Chemistry, London, 1997 Search PubMed.
- (a) R. N. Grimes, Carboranes, Academic Press, Elsevier Inc., Amsterdam, 2011 Search PubMed; (b) Boron Science: New technologies and Applications, ed. N. S. Hosmane, CRC Press, Boca Raton, FL, 2012 Search PubMed.
- (a) D. Grafstein and J. Dvorak, Inorg. Chem., 1963, 2, 1128–1133 CrossRef CAS; (b) R. A. Wiesboeck and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS; (c) M. F. Hawthorne, D. C. Young, P. M. Garrett, D. A. Owen, S. G. Schwerin, F. N. Tebbe and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 862–868 CrossRef CAS.
- Review: M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- (a) H. Tomita, H. Luu and T. Onak, Inorg. Chem., 1991, 30, 812–815 CrossRef CAS; (b) M. A. Fox, W. R. Gill, P. L. Herbertson, J. A. H. MacBride, K. Wade and H. M. Colquhoun, Polyhedron, 1996, 15, 565–571 CrossRef CAS; (c) M. A. Fox and K. Wade, Polyhedron, 1997, 16, 2517–2525 CrossRef CAS; (d) M. A. Fox and K. Wade, J. Organomet. Chem., 1999, 573, 279–291 CrossRef CAS; (e) J. Yoo, J.-W. Hwang and Y. Do, Inorg. Chem., 2001, 40, 568–570 CrossRef CAS; (f) J. A. Ioppolo, J. K. Clegg and L. M. Rendina, Dalton Trans., 2007, 1982–1985 RSC.
- (a) L. I. Zakharkin and V. N. Kalinin, Tetrahedron Lett., 1965, 6, 407–409 CrossRef; (b) F. Teixidor, C. Viñas, M. M. Abad, R. Nuñez, R. Kivekäs and R. Sillanpää, J. Organomet. Chem., 1995, 503, 193–203 CrossRef CAS; (c) M. A. Fox, A. E. Goeta, A. K. Hughes and A. L. Johnson, J. Chem. Soc., Dalton Trans., 2002, 2132–2141 RSC.
- (a) C. E. Willans, C. A. Kilner and M. A. Fox, Chem. – Eur. J., 2010, 16, 10644–10648 CrossRef CAS PubMed; (b) F. Zheng and Z. Xie, Dalton Trans., 2012, 41, 12907–12914 RSC.
- (a) A. S. Batsanov, R. C. B. Copley, M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard and K. Wade, J. Cluster Sci., 2006, 17, 119–137 CrossRef CAS PubMed; (b) Y. Taoda, T. Sawabe, Y. Endo, K. Yamaguchi, S. Fujii and H. Kagechika, Chem. Commun., 2008, 2049–2051 RSC.
- M. A. Fox, J. A. H. MacBride and K. Wade, Polyhedron, 1997, 16, 2499–2507 CrossRef CAS.
- M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard, A. Mackinnon, I. S. Neretin and K. Wade, Chem. Commun., 1999, 1649–1650 RSC.
- P. Dröse, C. G. Hrib and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 15540 CrossRef PubMed.
- Recent review articles: (a) F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS; (b) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC; (c) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS PubMed; (d) A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS PubMed; (e) A. A. Mohamed, H. E. Abdou and J. P. Fackler Jr, Coord. Chem. Rev., 2010, 254, 1253–1259 CrossRef CAS PubMed; (f) S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS PubMed; (g) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- (a) Z.-J. Yao, G. Su and G.-X. Jin, Chem. – Eur. J., 2011, 17, 13298–13307 CrossRef CAS PubMed; (b) Z.-J. Yao and G.-X. Jin, Organometallics, 2012, 31, 1767–1774 CrossRef CAS; (c) Z.-J. Yao, B. Xu, G. Su and G.-X. Jin, J. Organomet. Chem., 2012, 721–722, 31–35 CrossRef CAS PubMed; (d) Z.-J. Yao, Y.-J. Lin, Z.-H. Li and G.-X. Jin, Chem. – Eur. J., 2013, 19, 2611–2614 CrossRef CAS PubMed; (e) N. Harmgarth, D. Gräsing, P. Dröse, C. G. Hrib, P. G. Jones, V. Lorenz, L. Hilfert, S. Busse and F. T. Edelmann, Dalton Trans., 2014, 43, 5001–5013 RSC; (f) P. Hillebrand, C. G. Hrib, N. Harmgarth, P. G. Jones, V. Lorenz, M. Kühling and F. T. Edelmann, Inorg. Chem. Commun., 2014, 46, 127–129 CrossRef CAS PubMed.
- Recent reviews: (a) F. T. Edelmann, Z. Anorg. Allg. Chem., 2013, 639, 655–667 CrossRef CAS; (b) Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Hubert Schmidbaur on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Detailed experimental and analytical data as well as full crystallographic data for 4a, 4b, and 5. CCDC 1016818–1016820. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc06261b |
| This journal is © The Royal Society of Chemistry 2014 |