
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies towards the synthesis of halomon: asymmetric hexafunctionalisation of myrcene†
D. Christopher
Braddock
*,
Alison X.
Gao
,
Andrew J. P.
White
and
Mariko
Whyte
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.braddock@imperial.ac.uk; Fax: +44 (0)2075945805; Tel: +44 (0)2075945772
First published on 18th September 2014
Abstract
A novel dihydroxylation–dibromination–dihydroxylation sequence employing in situ protection of diols as boronate esters during the dihydroxylation reactions provides the first enantiomerically pure hexafunctionalised myrcene derivative. This concise four-step asymmetric sequence provides an advanced intermediate for the targeted synthesis of halomon via stereospecific transformations, where both stereogenic centres of the natural product have been set.
Halomon 1 is an acyclic pentahalogenated terpene isolated in pure form as a major component of the organic extracts of red algae Portieria hornemannii and characterised by X-ray crystallography by Boyd in 1992.1 Halomon's biological profile – initially found to display highly differential cytotoxicity against a diverse panel of human tumor cell lines,1 and also shown more recently to be an inhibitor of DNA methyl transferase 12 – has attracted much interest,3 but there is a lack of a reliable natural source from the algae due to site-to-site and temporal variations in terpene content.4 Synthetic efforts have resulted in the successful synthesis of halomon, but only in racemic form and as a mixture of stereoisomers, thereby requiring subsequent separation.5,6 There is therefore a clear need for an asymmetric and stereoselective strategy to access halomon and its related congeners4,7 for further biological evaluation. However, the introduction of each of the five halogens via site-selective functionalisations, including asymmetric fashioning of the two halogen-bearing stereocentres and installation of the vinyl chloride offers significant synthetic challenge.
Halomon 1 and its related congeners are considered to arise biogenetically via enzyme mediated Markovnikov addition of BrCl to the Δ1, Δ6 and/or C-3 methylene double bonds of myrcene 2 followed by elimination of HBr and/or HCl (Scheme 1, halomon only).8–10 With halomon 1 being isolated as a single enantiomer compound this implicates the intermediacy of enantiopure bromonium ions as promoted by bromoperoxidase in the algae.11 However, despite the recent rapid advance in asymmetric olefin halogenation methods,12 a general method for direct enantioselective bromonium ion formation on one face of an isolated alkene followed by intermolecular regioselective and stereospecific capture with a nucleophile does not yet exist,13 thus precluding attempted asymmetric synthesis of halomon 1 from myrcene 2 in this fashion.
 | ||
| Scheme 1 Halomon 1 and its proposed biogenesis from myrcene 2. | ||
On the other hand, we14 and Denmark15 have shown that enantiomerically pure bromonium ions can be generated from enantiopure bromohydrins and/or their O-derivatives. In particular, we have shown that either regioisomer of single enantiomer 1,2-bromotosylates of terminal aliphatic alkenes can generate single enantiomer bromonium ions that can be trapped with chloride anion to give enantiopure 1,2-chlorobromides.14a We have also shown that Sharpless dihydroxylation16 of a trisubstituted olefin provides access to single enantiomer bromohydrins via stereospecific epoxide formation17 and bromide ring-opening,18 where suitable subsequent activation leads to single enantiomer bromonium ions.14b,c With these considerations in mind, we have therefore targeted dibromotetrol (3R,6S,7S)-3 as an advanced intermediate for the synthesis of enantiopure halomon starting from myrcene 2 (Scheme 2). Here the diol of the original Δ6 trisubstituted alkene could potentially be advanced as previously demonstrated to an enantiopure bromonium ion and trapped with chloride anion. Likewise, the bisbromohydrin tetrad of the original Δ1 and C-3‡ methylene olefins of myrcene 2 could serve for the formation of two enantiomerically pure bromonium ions each to be trapped in a Markovnikov fashion with chloride anion. Herein, we report the successful asymmetric synthesis of dibromotetrol (3R,6S,7S)-3 from myrcene 2via a novel dihydroxylation–dibromination–dihydroxylation sequence employing in situ protection of diols as boronate esters during the dihydroxylation reactions. Critically, a citric acid buffered Upjohn reaction allows post-dibromination dihydroxylation to be effected successfully where other conditions compromise the diol dibromide products. Moreover the employment of a boronate ester as a protecting group allows quantitative and E-selective 1,4-dibromination of a diene that is otherwise impossible in the presence of a diol. To the best of our knowledge this represents the first synthesis of an enantiomerically pure hexafunctionalised myrcene derivative, and provides an advanced intermediate for the targeted synthesis of halomon 1via subsequent stereospecific transformations.
 | ||
| Scheme 2 Retrosynthesis of halomon 1 to myrcene 2via hexafunctionalised (3R,6S,7S)-dibromotetrol 3. | ||
Our studies commenced by using isoprene 4 as a model for the eastern portion of halomon, with a view to dihydroxylating known dibromide 5 into dibromodiol 6 (Table 1). However, we were aware that there have been no reports of a dihydroxylation of any kind on a 2-alkyl-1,4-dibromo-but-2-ene such as dibromide 5. Thus, isoprene was dibrominated according to the method of Alexakis19 giving the 1,4-dibromide 5 as a 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 E:Z mixture§¶ and dihydroxylation conditions were investigated (Table 1).
:6 E:Z mixture§¶ and dihydroxylation conditions were investigated (Table 1).
|
|
||
|---|---|---|
| Entry | Conditions | Yield 6a |
|
a Isolated yield after chromatography.
b Dihydroxylations were performed with 0.5 mol% K2OsO2(OH)4, 1 mol% (DHQD)2PHAL, K3Fe(CN)6 (3 equiv.), K2CO3 (3 equiv.) and MeSO2NH2 in tBuOH–H2O.
c Characteristic signals for epoxides could be seen in the 1H NMR of the crude reaction mixtures.
d An additional 1.5 mol% of K2OsO2(OH)4 and 3 mol% (DHQD)2PHAL were added, and a 3:2 ratio of K2CO3:NaHCO3 was employed.
e An additional 1 mol% of K2OsO2(OH)4 and 3 mol% (DHQD)2PHAL were added.
f The expected boronate ester was not observed.
g Acetone:water (8:1) as solvent.
h CH2Cl2 as solvent.
i No reaction of 5 was observed.
j
t
BuOH:H2O (1:1) as solvent.
|
||
| 1 | AD-mix β + MeSO2NH2b | 0c |
| 2 | AD-mix β + MeSO2NH2b + NaHCO3 | 0c |
| 3 | AD-mix β + MeSO2NH2b + NaHCO3d | 0c |
| 4 | AD-mix β + MeSO2NH2b + PhB(OH)2e | 0f |
| 5 | K2OsO2(OH)4, NMOg | 0c |
| 6 | K2OsO2(OH)4, NMO, PhB(OH)2h | 0i |
| 7 | K2OsO2(OH)4, NMO, citric acidj | 77 |
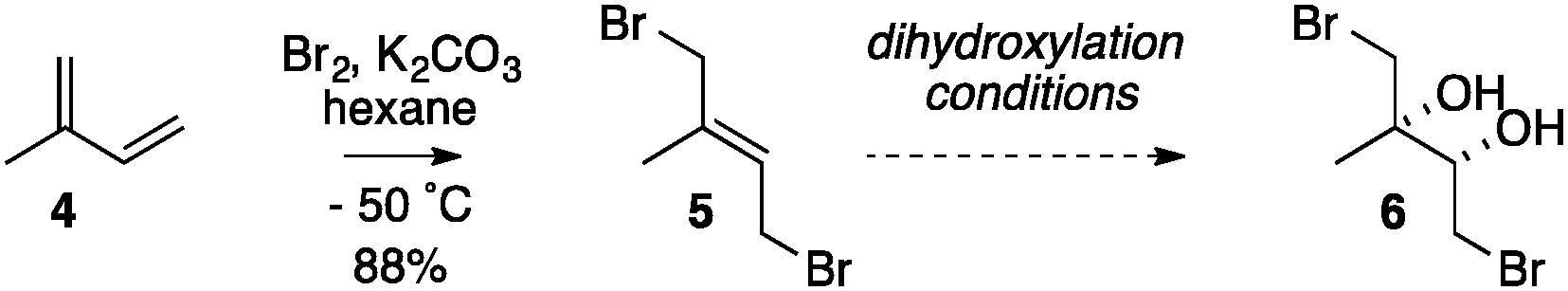
Attempted Sharpless asymmetric dihydroxylation (entry 1) of dibromide 5 with AD-mix-β and added methanesulphonamide16 failed, however, where the substrate was consumed but no diol 6 was detected, and characteristic signals for epoxides could be observed in the 1H NMR spectrum of the crude material.|| The limited literature precedent for osmium catalysed asymmetric dihydroxylation of allylic bromides suggests that NaHCO3-buffered AD-mix can suppress the unwanted pathways of hydrolysis of the starting material and base mediated ring-closure of the bromohydrin product to an epoxide.20 However, attempts to apply these conditions to the conversion of dibromide 5 into diol 6 also failed (entry 2). The use of other reported protocols for allylic bromides (entry 3)21 or attempted in situ capture of the diol as a boronate ester by addition of phenylboronic acid (entry 4)22 also failed. The observation of characteristic epoxide signals in each of the crude reaction mixtures led us to conclude that the inherently basic nature of the reaction medium was the central problem. Attempted non-asymmetric dihydroxylations under relatively neutral Upjohn conditions (entry 5)23 or using Narasaka's anhydrous modification of the Upjohn procedure (entry 6)24 also failed. Gratifyingly, the application of an osmium catalysed dihydroxylation under acidic conditions – a modified Upjohn reaction with NMO as the re-oxidant with added citric acid25 – allowed the smooth conversion of dibromide 5 into (±)-dibromodiol 6 in good isolated yield (entry 7),**†† and its structure was confirmed by X-ray crystallography.¶
Having demonstrated an effective dibromination–dihydroxylation on isoprene, attention turned to myrcene 2 where we wished to effect a dihydroxylation–dibromination–dihydroxylation sequence (cf.Scheme 2). A regioselective Sharpless dihydroxylation for the trisubstituted olefin of myrcene has been reported,26 but information on the sense of induction or enantiomeric excess was not provided. In the event, asymmetric dihydroxylation16 of myrcene 2 gave diol (3R)-727 as expected‡‡ in 91% ee §§(Scheme 3). Attempted dibromination of diene diol 7 to give dibromide 8 failed – as expected – due to presumed competing intramolecular bromoetherification pathways.¶¶ Instead, myrcene 2 was dihydroxylated with super AD-mix β28 and added phenyl boronic acid22 to give boronate ester 9 directly, also in 91% ee, as revealed by H2O2-mediated boronate deprotection24 to diol 7.§§ Here, not only does the addition of phenylboronic acid allow for the in situ protection of the newly installed 1,2-diol functionality, we also considered that the boronate ester would function as a uniquely effective protecting group in the subsequent bromination, where such diols otherwise protected in an additional synthetic step as e.g., benzylidenes or as alkyl acetals may retain sufficient nucleophilicity to participate in otherwise competing intramolecular bromoetherifications reactions.29
 | ||
| Scheme 3 Hexafunctionalisation of myrcene 2via a dihydroxylation–dibromination–dihydroxylation sequence employing boronate esters as protecting groups. | ||
Much to our delight, E-selective 1,4-dibromination of the diene 9 proceeded smoothly with molecular bromine without interference from the protected alcohol groups to give dibromide 10 in quantitative yield.|||| The application of the citric acid buffered Upjohn reaction25 (cf. dibromide 5 to diol 6, Table 1, entry 7) to dibromide 10 did not provide diol 11, but instead a moderate yield (45%) of inseparable bisboronates 12 and 13 was obtained. Here, under the conditions of reaction, evidently dihydroxylation is proceeding, but the isolation of the bisboronates implicates intermolecular boronate ester exchanges. Accordingly, citric acid buffered Upjohn dihydroxylation of 10 with added phenylboronic acid – a further novel variation on the Upjohn reaction – gave diastereomeric bisboronates 12 and 13 as an approximate 1:1 mixture of diastereoisomers in 85% yield.†† However, in contrast to the facile deprotection of boronate 9 with hydrogen peroxide, attempted global deprotection of the bisboronate mix 12 and 13 under the same conditions24 led only to low and variable mass recoveries (0–35% yield) of the two expected diastereomeric tetrols (3R,6R,7R)-3 and (3R,6S,7S)-3.*** Deprotection instead using modified conditions of Padwa,30 gave the separable diastereomeric dibromotetrols (3R,6S,7S)-3 and (3R,6R,7R)-3 in 82% combined yield.††† Dibromotetrol (3R,6S,7S)-3 proved to be crystalline, and the X-ray crystal structure (Fig. 1) confirmed the original sense of induction in the Sharpless asymmetric dihydroxylation as well as all other relative and absolute configurations.
 | ||
| Fig. 1 The crystal structure of (3R,6S,7S)-3. | ||
In conclusion we have demonstrated a successful asymmetric dihydroxylation–dibromination–dihydroxylation sequence employing in situ protection of diols as boronate esters during the dihydroxylation reactions to rapidly assemble the first enantiopure hexafunctionalised myrcene derivative in just four steps. This concise asymmetric sequence provides an advanced intermediate for the targeted synthesis of halomon 1via subsequent stereospecific transformations (vide supra),14 where both stereogenic centres of the natural product have already been set. Such work is ongoing and will be reported in due course.
We thank the Department of Chemistry, Imperial College London for support (to A. X. G. and M. W.).
Notes and references
- R. W. Fuller, J. H. Cardellina II, Y. Kato, L. S. Brinen, J. Clardy, K. M. Snader and M. R. Boyd, J. Med. Chem., 1992, 35, 3007–3011 CrossRef CAS.
- E. H. Andrianasolo, D. France, S. Cornell-Kennon and W. H. Gerwick, J. Nat. Prod., 2006, 69, 576–579 CrossRef CAS PubMed.
- (a) M. J. Egorin, D. L. Sentz, D. M. Rosen, M. F. Ballasteros, C. M. Kearns, P. S. Callery and J. L. Eiseman, Cancer Chemother. Pharmacol., 1996, 39, 51–60 CrossRef CAS; (b) M. J. Egorin, D. M. Rosen, S. E. Benjamin, P. S. Callery, D. L Sentz and J. L. Eiseman, Cancer Chemother. Pharmacol., 1997, 41, 9–14 CrossRef CAS PubMed.
- R. W. Fuller, J. H. Cardellina II, J. Jurek, P. J. Scheuer, B. Alvarado-Lindner, M. McGuire, G. N. Gray, J. R. Steiner, J. Clardy, E. Menez, R. H. Shoemaker, D. J. Newman, K. M. Snader and M. R. Boyd, J. Med. Chem., 1994, 37, 4407–4411 CrossRef CAS.
- T. Schlama, R. Baati, V. Gouverneur, A. Valleix, J. R. Falck and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2085–2087 CrossRef CAS.
- T. Sotokawa, T. Noda, S. Pi and M. Hirama, Angew. Chem., Int. Ed., 2000, 39, 3430–3432 CrossRef CAS.
- The racemic synthesis of two naturally occurring halogenated monoterpenes of the halomon class have been reported: M. E. Jung and M. H. Parker, J. Org. Chem., 1997, 62, 7094–7095 CrossRef CAS.
- B. J. Burreson, F. X. Wooland and R. E. Moore, Chem. Lett., 1975, 1111–1114 CrossRef CAS.
- Hirama and coworkers (ref. 6) employed a stepwise bromochlorination of myrcene to obtain 2,6-dibromo-3-(bromomethyl)-1,3,7-trichloro-7-methyloctane as a mixture of four diastereoisomers. However, all attempts to effect an elimination of HBr to yield halomon 1 were reported as unsuccessful.
- Geranyl diphosphate has been proposed to be the common biogenetic precursor for the wider family of monoterpenes in macrophytic red algae: M. L. Wise and R. Croteau, in Comprehensive Natural Product Chemistry, ed. D. H. R. Barton, K. Nakanishi and O. Meth-Cohn, Pergamon, Elmsford, NY, 1999, vol. 2, pp. 97–153 Search PubMed.
- For a review on halogenating enzymes see: A. Butler and M. Sandy, Nature, 2009, 460, 848–854 CrossRef CAS PubMed and references cited therein.
- For selected reviews see: (a) S. Zheng, C. M. Schienebeck, W. Zhang, H.-Y. Wang and W. Tang, Asian J. Org. Chem., 2014, 3, 366–376 CrossRef CAS; (b) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333–2343 RSC; (c) K. Murai and H. Fujioka, Heterocycles, 2013, 87, 763–805 CrossRef CAS PubMed; (d) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS PubMed; (e) U. Hennecke, Chem. – Asian J., 2012, 7, 456–465 CrossRef CAS PubMed; (f) C. K. Tan, L. Zhou and Y.-Y. Yeung, Synlett, 2011, 1335–1339 CAS; (g) A. Castellanos and S. P. Fletcher, Chem. – Eur. J., 2011, 17, 5766–5776 CrossRef CAS PubMed; (h) G. Chen and S. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306–8308 CrossRef CAS PubMed.
- For a review on asymmetric intermolecular halogenation of alkenes see: J. Chen and L. Zhou, Synthesis, 2014, 586–595 Search PubMed.
- (a) D. C. Braddock, S. A. Hermitage, L. Kwok, R. Pouwer, J. M. Redmond and A. J. P. White, Chem. Commun., 2009, 1082–1084 RSC; (b) D. C. Braddock, J. S. Marklew and A. J. F. Thomas, Chem. Commun., 2011, 47, 9051–9053 RSC; (c) D. C. Braddock, J. S. Marklew, K. M. Foote and A. J. P. White, Chirality, 2013, 25, 692–700 CrossRef CAS PubMed.
- S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232–1233 CrossRef CAS PubMed and references cited therein.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS.
- E. J. Corey, M. C. Noe and W.-C. Shieh, Tetrahedron Lett., 1993, 34, 5995–5998 CrossRef CAS.
- E. A. Couladouros and V. P. Vidali, Chem. – Eur. J., 2004, 10, 3822–3835 CrossRef CAS PubMed.
- C. A. Falciola, K. Tissot-Crosset, H. Reyneri and A. Alexakis, Adv. Synth. Catal., 2008, 350, 1090–1100 CrossRef CAS.
- H. C. Kolb, Y. L. Bennani and K. B. Sharpless, Tetrahedron: Asymmetry, 1993, 4, 133–141 CrossRef CAS.
- A Sharpless asymmetric dihydroxylation of a 1,6-dibromohexa-2,4-diene under modified conditions has been reported: O. Hidestål, R. Ding, A. Almesåaker and U. M. Lindström, Green Chem., 2005, 7, 259–261 RSC.
- C. H. Hövelmann and K. Muñiz, Chem. – Eur. J., 2005, 11, 3951–3958 CrossRef PubMed.
- V. VanRheenen, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 17, 1973–1976 CrossRef.
- A. Gypser, D. Michel, D. S. Nirschl and K. B. Sharpless, J. Org. Chem., 1998, 63, 7322–7327 CrossRef CAS PubMed and references cited therein.
- P. Dupau, R. Epple, A. A. Thomas, V. V. Fokin and K. B. Sharpless, Adv. Synth. Catal., 2002, 344, 421–433 CrossRef CAS.
- Dihydroxylation of myrcene using AD-mix α: J. Y. Wu, B. N. Stanzl and T. Ritter, J. Am. Chem. Soc., 2010, 132, 13214–13216 CrossRef CAS PubMed.
- (3R)-7 has been reported via an alternative route: H. Hioki, H. Ooi, M. Hamano, Y. Mimura, S. Yoshio, M. Kodama, S. Ohta, M. Yanai and S. Ikegami, Tetrahedron, 2001, 57, 1235–1246 CrossRef CAS.
- For ‘super’ AD-mix see footnote 76 in: H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- For recent representative examples of intramolecular bromoetherifications of unsaturated ethers see: (a) D. C. Braddock, D. S. Millan, Y. Perez-Fuertes, R. Pouwer, R. N. Sheppard, S. Solanki and A. J. P. White, J. Org. Chem., 2009, 74, 1835–1841 CrossRef CAS PubMed; (b) S. A. Synder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714–17721 CrossRef PubMed and references cited therein.
- A. Padwa and Q. Wang, J. Org. Chem., 2006, 71, 7391–7402 CrossRef CAS PubMed and reference cited therein.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterising data and copies of 1H and 13C NMR spectra for compounds 5–7, 9–10, 12/13 mix, (3R,6S,7S)-3 and (3R,6R,7R)-3; X-ray crystallographic details for 6 and (3R,6S,7S)-3. CCDC 1017807 and 1017806. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc06234e |
| ‡ However, to the best of our knowledge the formation of an enantiopure bromonium ion of a 1,1-disubstituted alkene has not yet been demonstrated. |
| § This ratio is comparable with that reported in literature at 92:8 (ref. 19). |
| ¶ See ESI† for details. |
| || The putative epoxides proved to be volatile and only traces of material could be isolated after attempted column chromatography. |
| ** Small quantities of the (R*,S*)-diol was also obtained (see ESI†). This arises from syn-dihydroxylation of the minor Z-dibromide 5. |
| †† The reaction mixture under these acidic conditions was characteristically green in colour that faded as the reaction proceeded. See ref. 25 for the significance of this colour. |
| ‡‡ The absolute configuration was assigned on the basis of the Sharpless mnemonic (ref. 16). |
| §§ The ee was calculated as 91% by comparison of the measured rotation with the reported rotation for (3R)-7 (ref. 27) (see ESI†). |
| ¶¶ Complex product mixtures were obtained. |
| |||| As a 96:4 E:Z mixture. |
| *** H2O2 deprotection studies with the phenyl boronate ester of dibromodiol 6 (not shown) showed characteristic signals for epoxides in the 1H NMR of the crude reaction mixtures. We invoke epoxide formation in the attempted H2O2 deprotection of boronates 12 and 13 also, and subsequent decomposition in the work-up and/or purification process (chromatography). |
| ††† The dibromotetrols were obtained as a ca. 1:1 mixture, and analytically pure samples of each could be obtained by careful column chromatography (see ESI†). |
| This journal is © The Royal Society of Chemistry 2014 |