
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of γ-hydroxynorvaline through combination of organo- and biocatalysis†
Robert C.
Simon
*a,
Eduardo
Busto
a,
Joerg H.
Schrittwieser
a,
Johann H.
Sattler
b,
Jörg
Pietruszka
c,
Kurt
Faber
a and
Wolfgang
Kroutil
*a
aDepartment of Chemistry, University of Graz, NAWI Graz, Heinrichstraße 28, A-8010-Graz, Austria. E-mail: wolfgang.kroutil@uni-graz.at; robert.simon@uni-graz.at
bAustrian Centre of Industrial Biotechnology (ACIB), Petersgasse 14, 8010 Graz, Austria
cInstitut für Bioorganische Chemie, Heinrich-Heine-Universität Düsseldorf im Forschungszentrum Jülich, Stetternicher Forst, Geb. 15.8, D-52426-Jülich, Germany
First published on 24th September 2014
Abstract
An efficient route for the synthesis of all four diastereomers of PMP-protected α-amino-γ-butyrolacton to access γ-hydroxynorvaline was established. The asymmetric key steps comprise an organocatalytic Mannich reaction and an enzymatic ketone reduction. Three reaction steps could be integrated in a one-pot process, using 2-PrOH both as solvent and as reducing agent. The sequential construction of stereogenic centres gave access to each of the four stereoisomers in high yield and with excellent stereocontrol.
Progressive legislative regulations with respect to sustainability, safety and quality improvements have changed the focus of contemporary chemical research to designing more economic processes. In this context, (asymmetric) catalysis has become the method of choice to replace stoichiometric amounts of reagents and auxiliaries, thereby facilitating an efficient and energy saving process.1 While each of the catalysis sub-fields (metal-, organo- and biocatalysis) has reached an impressive level of sophistication, efficiency can be further increased by combining methods from these different sub-fields. This allows exploiting and combining specific features of each type of catalyst in a highly cooperative fashion. For example, numerous reports have been published based on the combination of transition metals and organocatalysts;2 furthermore, biocatalytic transformations have been coupled with diverse metals.3 However, the combination of bio- and organocatalysts is still scarce.4,5
α-Amino-γ-butyrolactones are common structural elements in a series of natural products and pharmaceutical drugs that display diverse biological activities (cf.Fig. 1).6 Moreover, the corresponding amino acid (γ-hydroxynorvaline) has been isolated from natural sources7 and shows anti-diabetic properties.8 The wide-spread occurrence of the γ-butyrolactone unit has led to the development of a number of stereoselective syntheses mainly based on chemo-catalysis,9 while only few biocatalytic strategies have been developed.10 Most of the procedures either require multiple steps, are auxiliary-assisted, or based on ex-chiral pool precursors.11 Hence, access to only one or two out of four stereoisomers is provided. However, as the biological activity of bioactive compounds is related often to one specific unknown stereoisomer,12 a convenient access to all four diastereomers, e.g. for SAR-studies, is desired.
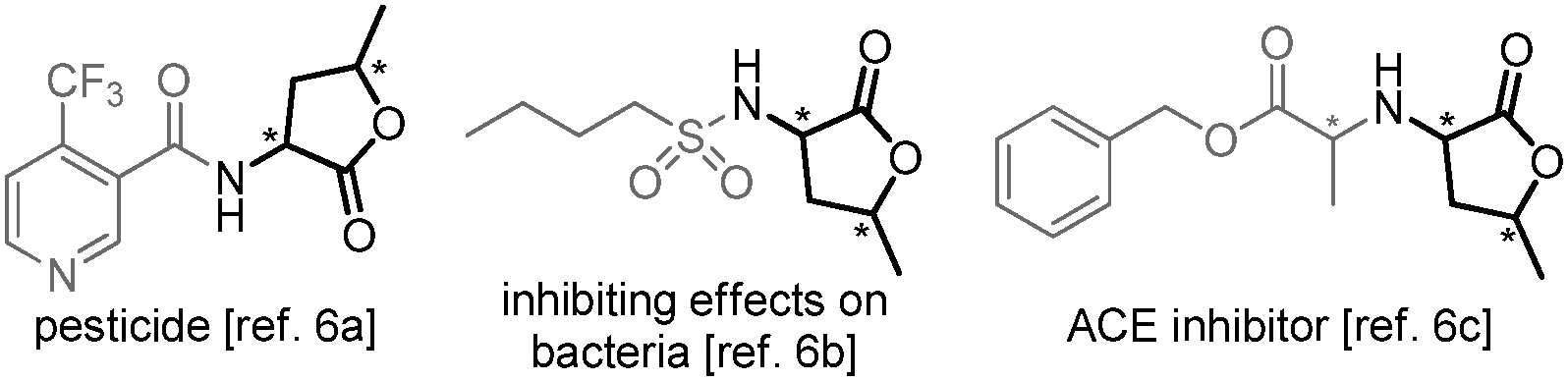 | ||
| Fig. 1 Selected bioactive compounds containing α-amino-γ-butyro-lactones. | ||
In this communication, a novel combination of organo- and biocatalysis is reported, which provides access to optically pure α-amino-γ-butyrolactones either in a step-wise fashion or in a cascade reaction.
In our study we focused initially on the bioreduction of racemic amino-ketoester rac-1, which is readily available by a proline catalysed Mannich reaction.13,14 A set of homo- and heterologously expressed alcohol dehydrogenases (ADHs)15 were assayed on analytical scale (5.6 mg, 20 mM) employing 2-propanol for NAD(P)+ regeneration. Asymmetric reduction of the carbonyl group of amino-ketoester 1 afforded the corresponding diastereomers of alcohol 2, which cyclised spontaneously to the desired 2-amino-lactones 3 (Scheme 1).
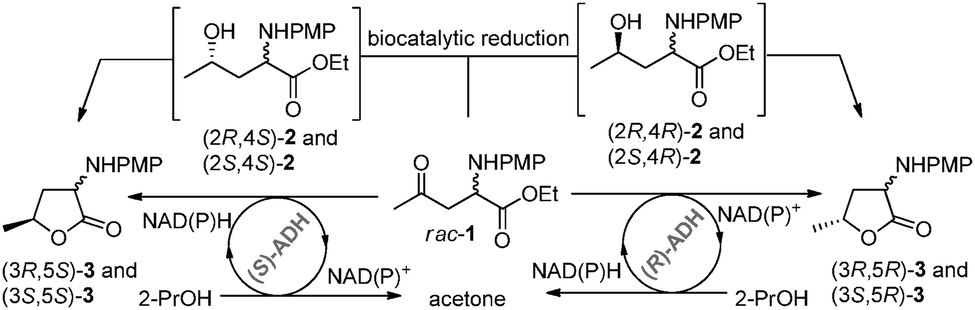 | ||
| Scheme 1 Alcohol dehydrogenase catalysed reduction of racemic ketoester rac-1 on analytical scale (20 mM, 5.6 mg). PMP = para-methoxyphenyl. | ||
A number of ADHs transformed rac-1 into the corresponding α-amino-lactones 3 with high conversion (selected results in Table 1; more details in the ESI†). For example, the (5S)-configured diastereomers (3S,5S)-3 and (3R,5S)-3 were accessible by the enzymes ADH-A15a (entry 1) and evo-1.1.03015b (entry 2), whereas the (5R)-diastereomers (3S,5R)-3 and (3R,5R)-3 were formed utilising evo-1.1.20015b (entry 5). The enzymes ADH-T15c (entry 3) and ADH-LS15d (entry 4) gave excellent results, too, with conversions of 83% and 86%, respectively. In all cases, the enantiomeric excess (ee) of all diastereomers of lactones 3 was >99%.
| Entry | Enzyme | Conv.b [%] | d.r. [syn/anti]b | eesyn3c [%] | eeanti3c [%] |
|---|---|---|---|---|---|
| a Reaction conditions: ketoester rac-1 (5.6 mg, 20 mM), KPi buffer (100 mM, pH 6.8), NAD(P)+ (1.0 mM), 10 vol% 2-PrOH, ADH (for units see ESI), 30 °C, 700 rpm on a thermoshaker (horizontal position), 24 h. b Determined via GC on an achiral stationary phase. c Determined via HPLC on a chiral stationary phase. | |||||
| 1 | ADH-A | 99 | 52/48 | >99 (3R,5S) | >99 (3S,5S) |
| 2 | evo-1.1.030 | 99 | 51/49 | >99 (3R,5S) | >99 (3S,5S) |
| 3 | ADH-T | 83 | 12/88 | >99 (3R,5S) | >99 (3S,5S) |
| 4 | ADH-LS | 86 | 71/29 | >99 (3S,5R) | >99 (3R,5R) |
| 5 | evo-1.1.200 | 99 | 52/48 | >99 (3S,5R) | >99 (3R,5R) |
| 6 | ADH-LB | 49 | 99/1 | >99 (3S,5R) | — |
| 7 | evo-1.1.270 | 49 | 99/1 | >99 (3S,5R) | — |
Among the enzymes tested, ADH-LB15e and evo-1.1.27015b gave unexpected results as in both cases mainly one diastereomer (3S,5R)-3 was detected (entries 6 and 7). Since the conversions remained below 50%, we assumed a selective kinetic resolution with respect to the existing stereogenic centre in β-position to the carbonyl group. In order to gain a better understanding of such a rare case of distant stereorecognition by ADHs,16 further investigations were performed: racemic and enantiopure Mannich-bases rac-/(R)-/(S)-1 were incubated with ADH-LB and the reaction progress was monitored over time (Fig. 2). Thereby it was found that rac-1 gave the (3S,5R)-3 diastereomer as sole product with 40% conversion after eight hours, while (R)-1 was not transformed. The pure enantiomer (S)-1 reacted fastest, reaching 70% conversion within the same time.
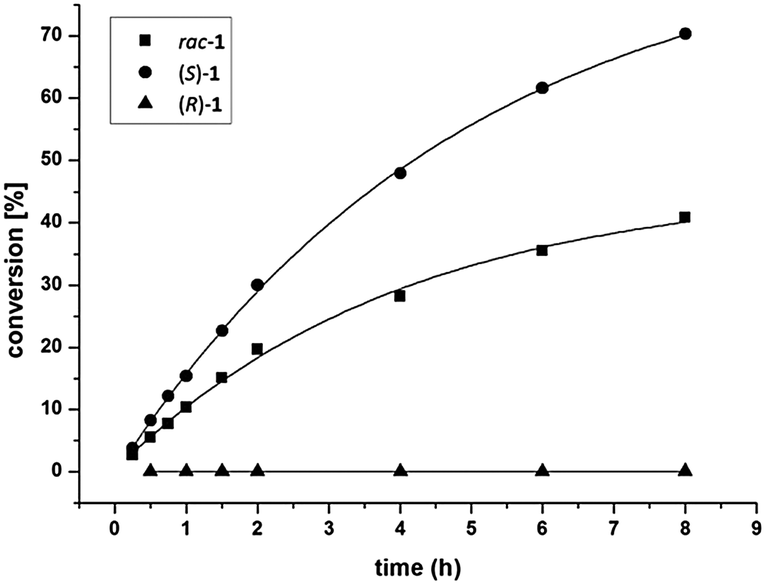 | ||
| Fig. 2 Time course of the biocatalytic reduction of racemic and enantiopure Mannich-base 1 with the enzyme ADH-LB (double determination). | ||
The experimental observations were additionally supported by in silico docking studies: only substrate (S)-1 was found to fit in the active site enabling a productive binding mode; the substrate occupies therein a pro-R conformation in which the carbonyl group can be activated by two hydrogen bonds through amino acids Tyr152 and Ser142 (Fig. 3). Attempts to dock the opposite enantiomer (R)-1 into the active site failed to provide a reasonable productive binding mode (see ESI†). Hence, the structure of ADH-LB allows a chiral recognition of a remote stereocentre enabling a kinetic resolution.16 Attempts to couple the diastereoselective reduction of 1 with a racemisation of 1 in a dynamic kinetic resolution failed so far due spontaneous degradation of 1via elimination of anisidine over time.
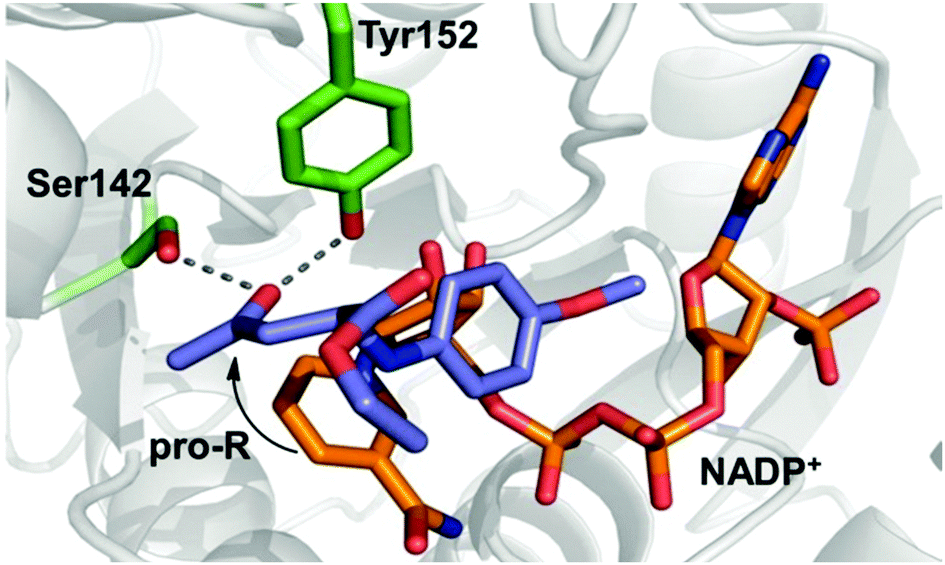 | ||
| Fig. 3 Docking study: productive binding mode of Mannich-base (S)-1 (purple) and the cofactor NADP+ (orange) in the active site of ADH-LB. | ||
The next step was the synthesis of all four stereoisomers of amino-lactone 3; for this purpose, the biocatalysed reduction of ketoester 1 was optimised using alcohol dehydrogenases ADH-A and evo-1.1.200. Investigating 2-PrOH as a cosolvent revealed that a substrate concentration of 30 mM (7.95 mg mL−1, 10 vol% 2-PrOH) led to completion of the reaction in the case of the (S)-selective ADH-A within 24 h (for details, see ESI†). The (R)-selective enzyme evo-1.1.200 transformed even 50 mM (13.3 mg mL−1, 30 vol% 2-PrOH) to completion within 24 h. The optimum pH was determined to be 6.3 in order to keep the partial racemisation of the optically pure Mannich adduct 1 at a minimum (see ESI†). In the subsequent semi-preparative scale transformations [56 mg (S)-1] >99% conversion was achieved; however, ring closure was incomplete, as mainly the optically pure amino-alcohol 2 rather than the lactone 3 was isolated (ca. 50–70%; see ESI†).17 Assuming that the corresponding methyl ester of 1 would undergo faster lactonisation than the ethyl analogue, a transesterification step (HCl–MeOH) was integrated into the reaction sequence. The optimised conditions finally allowed conversion of the pure amino-ester (R)-/(S)-1 (up to >300 mg), readily accessible by a proline-catalysed Mannich reaction of acetone and imine 4,14 into the corresponding lactones 3. The syn- and anti-diastereomers were isolated in excellent yields (78–82%) and with remarkable stereocontrol (de >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and ee >99% in all cases; see Scheme 2).
:2 and ee >99% in all cases; see Scheme 2).
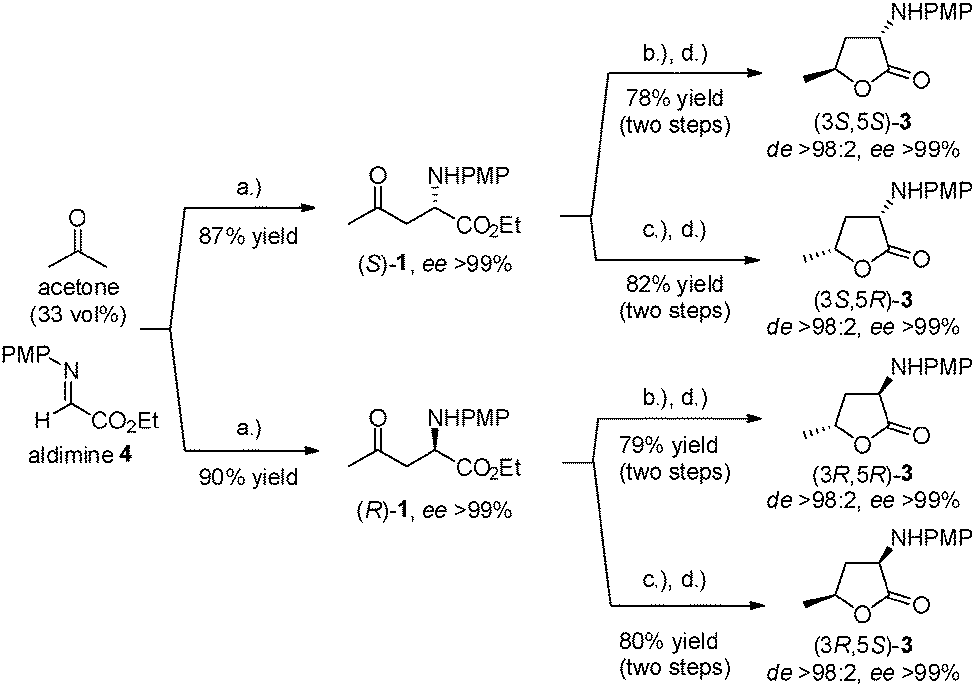 | ||
| Scheme 2 Synthesis of all four diastereomers of PMP-protected α-amino-γ-butyrolactone 3 through combination of organo- and biocatalysis. Reagents and conditions: (a) D-/L-proline (0.25 eq.), DMSO, 3 h, rt; (b) (R)-/(S)-1 (30 mM), ADH-A, NAD+, buffer (100 mM, pH 6.3), 2-PrOH (10 vol%), 30 °C, 24 h; (c) (R)-/(S)-1 (50 mM), evo-1.1.200, NAD+, buffer (100 mM, pH 6.3), 2-PrOH (30 vol%), 30 °C, 24 h; (d) HCl·Et2O (2.0 eq.), MeOH, 16 h, rt. PMP = para-methoxyphenyl. | ||
As an alternative to the conventional step-by-step synthesis (Scheme 2) two sequential approaches were established. In a first approach, aldimine 4 was formed in 2-PrOH, which also served as a solvent for the subsequent Mannich reaction and as the reducing agent for the enzyme-mediated reduction (after dilution with buffer solution). All three steps were accomplished in one pot, after which the transesterification-lactonisation cascade was performed in a separate vessel. The whole sequence afforded the syn-configured lactone (3S,5R)-3 in a remarkable 47% yield with a d.r. of 86/14 in favour of the syn-diastereomer (Scheme 3a: 5 steps in total, 2 pots, 90% average yield per step). The diminished d.r. in comparison to the previous results can be rationalised by the usage of 2-PrOH for the proline-catalysed reaction, which under these conditions afforded (S)-1 with 72% ee.
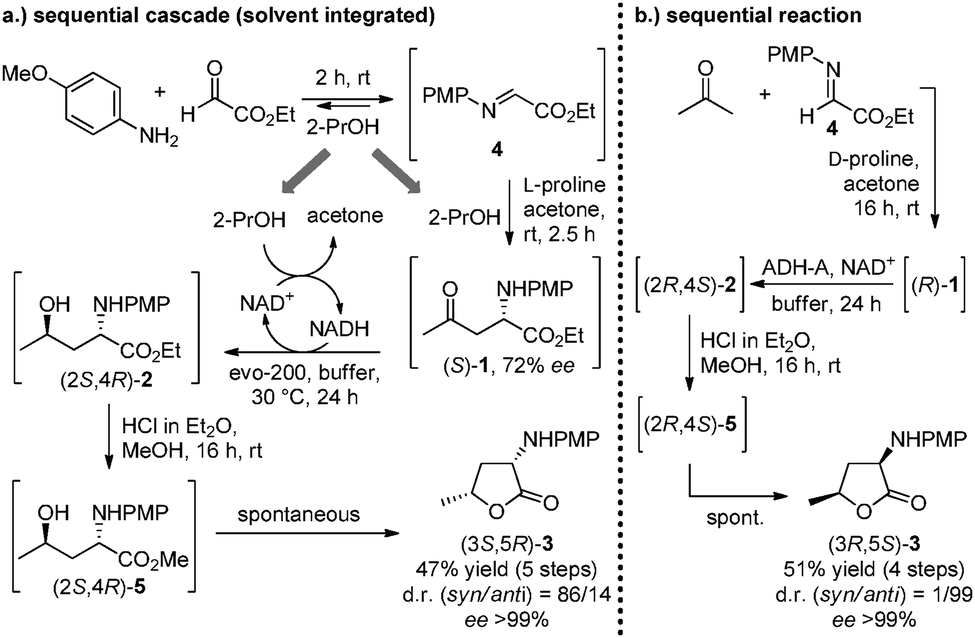 | ||
| Scheme 3 Synthesis of α-amino-γ-butyrolactones by means of two different approaches: (a) sequential cascade reaction in which the solvent (2-PrOH) was integrated in the first three steps (5 steps, 2 pots); (b) conventional sequential reaction sequence with the preformed aldimine (4 steps, 2 pots). | ||
In a second approach, the Mannich reaction was carried out with the preformed aldimine 4 in acetone as solvent, and amino-ketoester (R)-1 was obtained at prolonged reaction time (16 h) with >99% conversion and in optically pure form (ee >99%). After evaporation of the solvent, the crude product was subjected to reduction by ADH-A in buffer–2-PrOH. The final transesterification-lactonisation cascade furnished the diastereomerically pure amino lactone (3R,5S)-3 in 51% yield with an excellent d.r. of 99:1 (Scheme 3b: 4 steps, 2 pots, 85% average yield per step).
Final PMP-deprotection of the diastereomerically pure α-amino-lactone 3 was achieved employing TCCA (trichloroisocyanuric acid) in MeOH,18b providing the hydrochloride salt of 6. Unexpectedly, other oxidative deprotection methods [PhI(OAc)2, CAN, HIO6, laccase, etc.]18 were unsuccessful. Intensive optimisation of the purification of 6 with respect to the solvent, acid, reaction time and work-up procedure finally provided access to γ-hydroxynorvaline: the natural product was isolated diastereomerically pure after a base promoted ring opening (58% yield over two steps) as demonstrated for the (2R,4S)-diastereomer (Scheme 4).
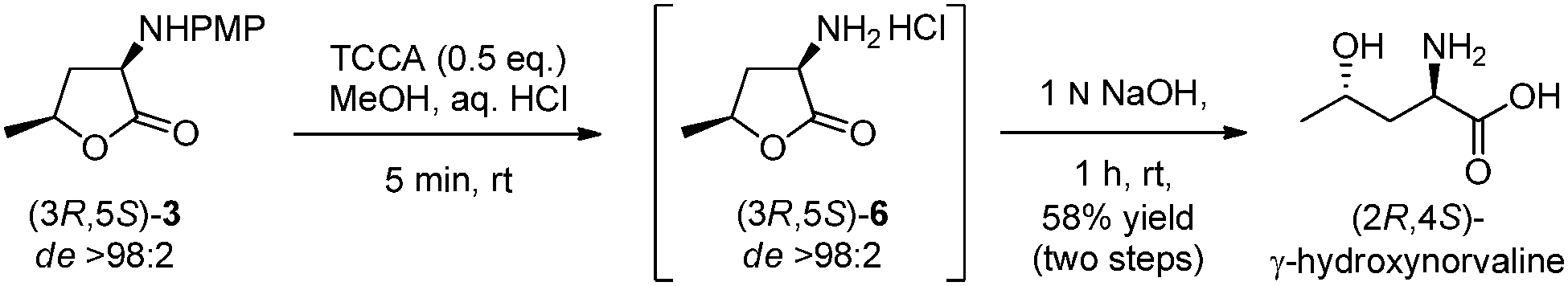 | ||
| Scheme 4 PMP deprotection to yield natural γ-hydroxynorvaline. | ||
In conclusion, a straightforward synthesis of all four diastereomers of PMP-protected α-amino-γ-butyrolactone to access γ-hydroxynorvaline has been established. The sequential construction of the stereogenic centres was accomplished by combination of organo- and biocatalysed key steps. In one approach, three sequential steps including the organo- as well as the biocatalysed transformations were performed in one pot, integrating 2-propanol first as solvent and then as reducing agent.
E. B. received funding from the European Commission by a Marie Curie Actions-Intra-European Fellowship (IEF) in the project “BIOCASCADE” (FP7-PEOPLE-2011-IEF). COST Action CM1303 “Systems Biocatalysis” is acknowledged. J.H.S. has been supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET-Funding Program.
Notes and references
- R. Noyori, Nat. Chem., 2009, 1, 5 CrossRef CAS PubMed.
- (a) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC; (b) M. Á. Fernández-Ibañez, B. Maciá, D. A. Alonso and I. M. Pastor, Molecules, 2013, 18, 10108 CrossRef PubMed; (c) L. Stegbauer, F. Sladojevich and D. J. Dixon, Chem. Sci., 2012, 3, 942 RSC; (d) C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 2999 CrossRef CAS.
- (a) B. Martín-Matute and J.-E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226 CrossRef PubMed; (b) O. Pàmies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247 CrossRef PubMed.
- For a recent review see: H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171 CrossRef PubMed.
- (a) K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2009, 48, 9355 CrossRef CAS PubMed; (b) G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944 CrossRef CAS PubMed; (c) G. Rulli, M. Heidlindemann, A. Berkessel, W. Hummel and H. Gröger, J. Biotechnol., 2013, 168, 271 CrossRef CAS PubMed; (d) M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099 CrossRef CAS; (e) A. Kinnell, T. Harman, M. Bingham, A. Berry and A. Nelson, Tetrahedron, 2012, 68, 7719 CrossRef CAS PubMed; (f) R. Millet, A. M. Träff, M. L. Petrus and J.-E. Bäckvall, J. Am. Chem. Soc., 2010, 132, 15182 CrossRef CAS PubMed.
- (a) W. Schaper, M. Beckmann, U. Doller, G. Krautstrunk, D. Jans, W. Hempel and J. M. Waibel, US2004/6047 A1, US, 2004; (b) J. Nielsen and M. Givskov, WO03106445 (A1), 2003; (c) H. Hasegawa, N. Shiori, T. Narita and T. Katori, US4876359 A1, 1989.
- (a) L. Fowden, Nature, 1966, 209, 807 CrossRef CAS; (b) P. Matzinger, P. Catalfomo and C. H. Eugster, Helv. Chim. Acta, 1972, 55, 1478 CrossRef CAS.
- (a) G. Ribes, C. Broca, P. Petit, M. Jacob, Y. Baissac, M. Manteghatti, M. Roye and Y. Sauvaire, Diabetologia, 1996, 39, A234 Search PubMed; (b) C. Broca, M. Manteghetti, R. Gross, Y. Baissac, M. Jacob, P. Petit, Y. Sauvaire and G. Ribes, Eur. J. Pharmacol., 2000, 390, 339 CrossRef CAS.
- For a review see: M. Seitz and O. Reiser, Curr. Opin. Chem. Biol., 2005, 9, 285 CrossRef CAS PubMed.
- (a) S. E. Franz, R. R. Watkins, L. A. Wright, B. A. Weaver, R. C. Hartage, I. Ghiviriga, G. Gumina and B. D. Feske, Synthesis, 2013, 2171 CAS; (b) T. Classen, M. Korpak, M. Schölzel and J. Pietruszka, ACS Catal., 2014, 4, 1321 CrossRef CAS; (c) M. Korpak and J. Pietruszka, Adv. Synth. Catal., 2011, 353, 1420 CrossRef CAS; (d) A. Díaz-Rodríguez, W. Borzęcka, I. Lavandera and V. Gotor, ACS Catal., 2013, 4, 386 CrossRef.
- (a) J. Ariza, J. Font and R. M. Ortuño, Tetrahedron, 1990, 46, 1931 CrossRef CAS; (b) L. J. Drummond and A. Sutherland, Tetrahedron, 2010, 66, 5349 CrossRef CAS PubMed; (c) M. Jacob, M. L. Roumestant, P. Viallefont and J. Martinez, Synlett, 1997, 691 CrossRef CAS PubMed; (d) C. Schmeck and L. S. Hegedus, J. Am. Chem. Soc., 1994, 116, 9927 CrossRef CAS; (e) M. Sendzik, W. Guarnieri and D. Hoppe, Synthesis, 1998, 1287 CrossRef CAS PubMed; (f) M. D. Swift and A. Sutherland, Org. Biomol. Chem., 2006, 4, 3889 RSC.
- (a) W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760 CrossRef CAS; (b) B. K. Patel and A. J. Hutt, in Chirality in Drug Design and Development, ed. I. K. Reddy and R. Mehvar, New York, 2004 Search PubMed.
- For reviews on organocatalysed Mannich reactions see: (a) X. Cai and B. Xie, ARKIVOC, 2013, 264 CrossRef CAS; (b) J. M. M. Verkade, L. J. C. v. Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC; (c) A. Córdova, Acc. Chem. Res., 2004, 37, 102 CrossRef PubMed.
- For pioneering work in the organocatalysed Mannich reaction see: (a) B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827 CrossRef CAS PubMed; (b) A. Córdova, W. Notz, G. Zhong, J. M. Betancort and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1842 CrossRef PubMed; (c) A. Córdova, S. Watanabe, F. Tanaka, W. Notz and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1866 CrossRef PubMed; (d) A. Córdova and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 7749 CrossRef; (e) Y. Hayashi, W. Tsuboi, I. Ashimine, T. Urushima, M. Shoji and K. Sakai, Angew. Chem., Int. Ed., 2003, 42, 3677 CrossRef CAS PubMed; (f) I. Ibrahem and A. Córdova, Chem. Commun., 2006, 1760 RSC; (g) W. Zhang, S. Saaby and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 4476 CrossRef PubMed.
- ADHs employed during the studies: ADH-A originates from Rhodococcus ruber; see: (a) K. Edegger, C. C. Gruber, T. M. Poessl, S. R. Wallner, I. Lavandera, K. Faber, F. Niehaus, J. Eck, R. Oehrlein, A. Hafner and W. Kroutil, Chem. Commun., 2006, 2402 RSC; (b) Evozymes were obtained from Evocatal (origin not stated); (c) ADH-T was obtained from Codexis Inc. (origin not stated); (d) ADH-LS originates from Leifsonia sp.; see K. Inoue, Y. Makino and N. Itoh, Appl. Environ. Microbiol., 2005, 3633 CrossRef CAS PubMed; (e) ADH-LB originates from Lactobacillus brevis; see: W. Hummel and B. Riebel, Biotechnol. Lett., 2003, 25, 51 CrossRef CAS.
- Only two related recent reports were found about a kinetic resolution with respect to a distant stereogenic centre: (a) V. Höllrigl, K. Otto and A. Schmid, Adv. Synth. Catal., 2007, 349, 1337 CrossRef; (b) F. Hollmann, A. Kleeb, K. Otto and A. Schmid, Tetrahedron: Asymmetry, 2005, 16, 3512 CrossRef CAS PubMed ; for a review on recognition of remote stereogenic centers see: ; (c) M. A. Blasco and H. Gröger, Bioorg. Med. Chem., 2014 DOI:10.1016/j.bmc.2014.06.024.
- The ring-closure was found to be promoted by the GC injector pretending full conversion to the lactone 3. In an additional control experiment the purified alcohol 2 was subjected to GC and GC-MS analysis, showing only the signal for the lactone 3.
- (a) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, H. E. Schoemaker, M. Schürmann, F. L. van Delft and F. P. J. T. Rutjes, Adv. Synth. Catal., 2007, 349, 1332 CrossRef CAS; (b) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, P. L. Alsters, F. L. van Delft and F. P. J. T. Rutjes, Tetrahedron Lett., 2006, 47, 8109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of each experiment, full characterisation of all compounds and copy of HPLC traces and NMR data. See DOI: 10.1039/c4cc06230b |
| This journal is © The Royal Society of Chemistry 2014 |