
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An optimised scalable synthesis of H2O@C60 and a new synthesis of H2@C60†
Andrea
Krachmalnicoff
,
Malcolm H.
Levitt
and
Richard J.
Whitby
*
Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: rjw1@soton.ac.uk
First published on 9th September 2014
Abstract
New high-yielding synthetic routes to the small-molecule endofullerenes H2O@C60, D2O@C60 and H2@C60 are described. The use of high temperatures and pressures for the endohedral molecule incorporation are avoided. A new partial closure step using PPh3, and final suturing using a novel Diels–Alder/retro-Diels–Alder sequence are amongst the advances reported.
The molecular structure of C60 fullerene consists of a carbon cage enclosing a spherical cavity with a diameter of approximately 3.5 Å. Small-molecule endofullerenes encapsulate a single small molecule such as H2 or H2O within the cavity.1,2 The encapsulated molecule behaves as a confined quantum rotor, displaying a rich energy level structure,3 which may be studied by neutron scattering,4,5 infrared spectroscopy5,6 and nuclear magnetic resonance (NMR).3,5,7 The stability and homogeneity of endofullerenes such as H2@C60 and H2O@C60 allows the convenient and precise study of important physical phenomena such as nuclear spin isomer conversion.5,7,8 It is possible to detect the endohedral interconversion of ortho and para-water, and to show that the conversion is a bimolecular process even under cryogenic conditions, with neighbouring pairs of ortho-water molecules interacting so as to generate a pair of para-water molecules.7 Apart from their intrinsic interest, such observations may underpin new routes to the generation of nuclear hyperpolarization, analogous to the chemical reactions of para-enriched hydrogen gas.9 The high demand for these unique substances has highlighted limitations of the current methods for their preparation. The synthetic route to endofullerenes has been described as “molecular surgery”10 and involves three stages; (i) opening and progressive widening of an orifice in the C60 cage through exohedral reactions, (ii) introduction of a small molecule through the orifice into the cavity, (iii) further reactions leading to reclosure of the C60 cage and encapsulation of the endohedral molecule. The final (reclosure) stage is the most difficult and has been accomplished only by Komatsu and Murata using two different routes leading to H2@C60 (8 steps, 8% overall yield),1 and H2O@C60 (6 steps, 5% yield) respectively.2 Both syntheses require specialised high pressure equipment for the endohedral molecule incorporation step. Herein we report more efficient and practical routes to these important molecular systems.
The key feature of the H2O@C60 synthesis is that the open-cage derivative 1, having a 13-membered orifice, can be converted in situ into its dehydrated form 2 that has a 16-membered orifice. Compound 2 is able to incorporate a water molecule but is easily re-hydrated, reducing once again the size of the orifice trapping the guest molecule inside the cage. Compound H2O@1 is then reduced with (iPrO)3P to H2O@3 which in turn is pyrolysed under vacuum to produce H2O@C60 (Scheme 1).
 | ||
| Scheme 1 H2O@C60 synthesis.2 | ||
The reported route2 to the open fullerene 1 worked well but we lacked the equipment to apply the very high pressures (9000 atm) used to efficiently encapsulate the water molecule. It was already known that compound 1 could incorporate water (8%) under atmospheric pressure2 and that higher filling factors could be achieved under similar conditions in other open fullerenes bearing larger orifices.11 Published studies on the filling of hydrophobic cavities by water molecules suggested that an increased filling factor could be as well achieved by changing variables other than pressure.12 On these premises we investigated the filling of 1 under low pressure conditions.
Heating a solution of 1 in toluene in an NMR tube in the presence of water (5.6 equiv.) at 120 °C reached a maximum of 23% incorporation after 36 h reflecting the equilibrium situation. Increasing reaction time further gave substantial decomposition. Changing to a sealed tube which could be uniformly heated to prevent condensation in cooler regions increased incorporation to 45%. Using a large excess of water did not significantly change incorporation. Changing to other aromatic solvents demonstrated increased incorporation (benzene, 39%; ortho-dichlorobenzene, 61%; 1-chloronapthalene, 67%). Using 1-chloronapthalene as the solvent and lowering the temperature to 100 °C gave 78% H2O incorporation after 48 h providing a practical alternative to high pressure procedures. The same process can be extended to the synthesis of D2O@1 but to avoid contamination with HDO@1 and H2O@1 it was necessary to pre-form the dehydrated compound 2 by refluxing a toluene solution of 1 for one hour, passing the condensed solvent through a column of activated 3 Å molecular sieves. Subsequent removal of solvent to give 2, dissolution in 1-chloronapthalene, addition of D2O and heating as above gave D2O@1 with no HOD@1 or H2O@1 visible by 1H NMR spectroscopy. Compound 2 was recently isolated using a different protocol.13
In an effort to complete the synthesis of H2O@C60 our attention turned to suturing H2O@1. Unfortunately when compound H2O@1 was reacted with (iPrO)3P the expected product H2O@3 was isolated in only 34% yield. The same reaction mixture yielded 31% of the novel compound H2O@4. The structure of the latter can be deduced from the appearance in the 1H NMR spectrum of a singlet at δ 7.80 ppm from the proton present on the rim of the orifice and a broad signal at δ 5.21 attributable to the single hydroxyl group. The DFT-GIAO calculated spectra suggest the formation of regioisomer 4. Compound 4 was the only product isolated (76% yield) when 1 was reacted with (iPrO)3P at room temperature. Compound 4 is inert to (iPrO)3P and H2O@4 does not lose its endohedral water molecule upon heating. A somewhat similar reactivity towards alkyl phosphites has been reported for another open-cage fullerene, and the mechanism of the reduction studied theoretically.14 Pre-formed 2 reacted with (iPrO)3P at room temperature to afford compound 3 in 33% yield without contamination by 4. It is likely that under the reported closure conditions2 it is a mixture of H2O@1 and H2O@2 formed in situ which are reacting with (iPrO)3P to give varying mixtures of H2O@4 and H2O@3 (Scheme 2).
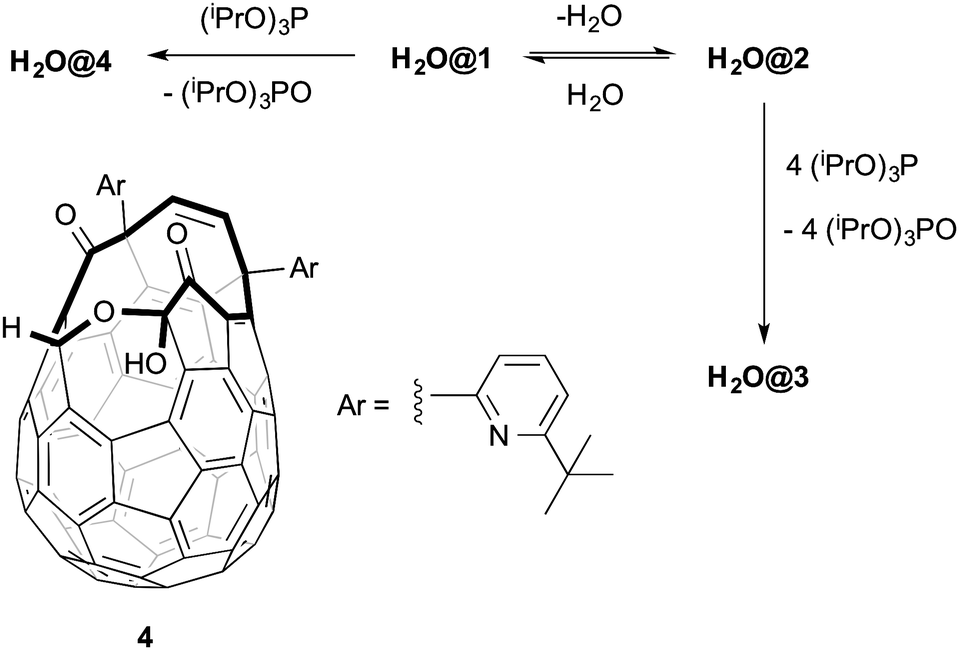 | ||
| Scheme 2 Reactivity of H2O@1 and H2O@2 towards triisopropyl phosphite. | ||
In order to overcome this side reactivity of H2O@1 towards alkyl phosphites we looked at alternative reagents and found that reaction with excess Ph3P at 120 °C cleanly gave compound H2O@5, isolated in 84% yield. Compound H2O@4 was not formed and, even with longer reaction times, reduction to compound H2O@3 was not observed. No endohedral water was lost in the formation of H2O@5 for which this reaction provides the first synthesis. Compound H2O@5 could be reacted with excess (iPrO)3P to give H2O@3 which was isolated after chromatography in 84% yield. In comparison with the reported direct reaction with (iPrO)3P,2 the yield of H2O@3 afforded in the two step process, is higher overall (69%) (Scheme 3).
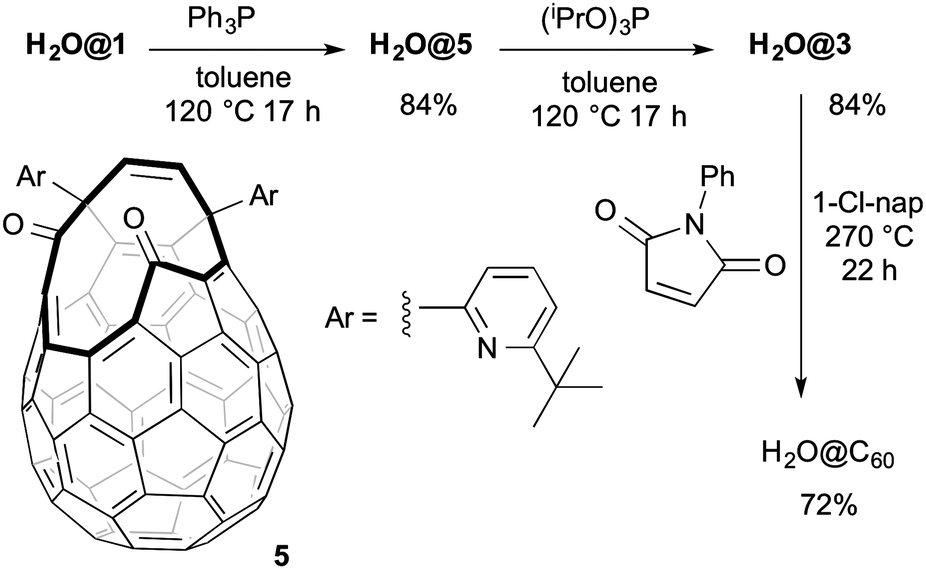 | ||
| Scheme 3 Optimised suturing procedure of compound H2O@1 to give H2O@C60. | ||
Our attention finally turned to the last step of the closure – the conversion of H2O@3 into H2O@C60. The reported yield of 29% was achieved by vacuum pyrolysis (360 °C) of H2O@3 dispersed on dry neutral alumina.2 Unfortunately in our hands we could isolate only traces of the product. A reasonable mechanism involves an initial [4+2] intramolecular cycloaddition to give strained intermediate 6 which rearranges via radical cleavage (formally a retro-[4+4] cycloaddition) of the strained C–C cyclopropane bonds generating intermediate 7. Finally, a retro-[2+2+2] cycloaddition leads to the extrusion of the side aromatic groups and formation of the C60 structure (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism for the conversion of 3 into C60. The C60 skeleton other than the orifice carbons (solid line) has been omitted for clarity. | ||
The last step (7 to C60) is likely to have the highest activation energy. We postulated that a lower energy reaction profile could be followed in presence of N-phenylmaleimide, a strong dienophile that was expected to react readily with the intermediate 7 in a [4+2] cycloaddition to afford adduct 8 which is a good substrate for a retro [4+2] cycloaddition to regenerate the C60 structure. These predictions were confirmed experimentally by reacting compound H2O@3 with N-phenylmaleimide in 1-chloronaphthalene under reflux conditions. After 20 hours a clean conversion to H2O@C60, was observed and column chromatography afforded the product in 90% yield. Further purification by sublimation gave pure H2O@C60 in 72% overall yield (Scheme 3). The same route has been followed to complete the synthesis of D2O@C60 in identical yield.
Komatsu has reported the synthesis of H2@C60 in 8 steps and 8% overall yield via insertion of H2 into a different cage-opened fullerene at 800 atm and 200 °C.1 Given the efficiency of the opening/closure described above a more convenient synthesis seemed possible. Incorporation of molecular hydrogen into 2 (formed in situ from 1 using 3 Å molecular sieves) at 120 °C and 120 atm H2 for 20 h gave H2@1 after work-up with a 60% filling factor. The lower temperature (120 °C cf. 200 °C) allowed reasonable incorporation at moderate pressures. It is likely that we would obtain complete filling at the high pressures (800 atm) used previously.1
In order to test if the system could be closed without the loss of the endohedral hydrogen molecule, a mixture of H2O@1 and H2@1 was reacted with (iPrO)3P in refluxing toluene. All the endohedral hydrogen was lost while all the water was retained inside. The problem was overcome by carrying out the phosphite reaction under hydrogen pressure. Thus compound 1 was heated to 120 °C in o-dichlorobenzene under hydrogen pressure (120 atm) in the presence of molecular sieves to give H2@2 (60% H2 incorporation). The bomb was then depressurised and (iPrO)3P or Ph3P was added before pressurising with hydrogen again and heating to 120 °C overnight. The reactions afforded 60% filled H2@3 in 50% yield and 60% filled H2@5 in 84% yield respectively. Both H2@3 and H2@5 can be reacted in an analogous way as already described for H2O@3 and H2O@5 to isolate 60% filled H2@C60 (Scheme 5).
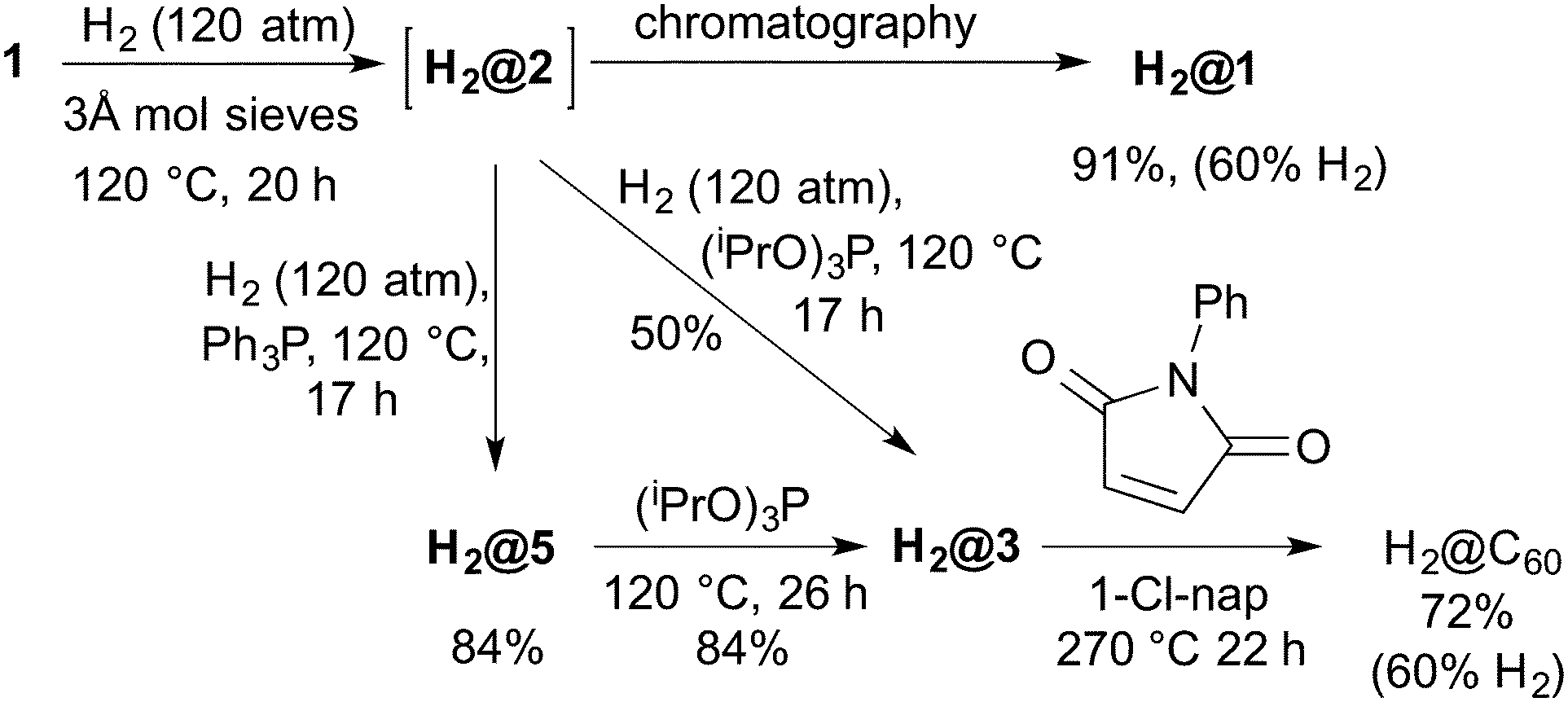 | ||
| Scheme 5 Synthesis of H2@C60 from 1. | ||
In conclusion a systematic study of the H2O@C60 synthesis developed by Murata2 has been carried out. It has been demonstrated that 78% incorporation of H2O and D2O into compound 1 can be achieved without the need for high pressure equipment. Treatment of hemiacetal 1 with (iPrO)3P at room temperature gave the reduced product 4 and H2O@4 was a major by product during the (iPrO)3P induce closure of H2O@1 to give H2O@3. Dehydration of hemi-acetal 1 to give tetraketone 2 before reaction with (iPrO)3P at room temperature gave 3 without formation of 4. A new partial closure procedure from H2O@1 using PPh3 allowed H2O@5 to be isolated for the first time, and subsequent deoxygenation with (iPrO)3P gave H2O@3 in improved overall yield. A new method for closing the fullerene H2O@3 to afford H2O@C60 was developed using a Diels–Alder/retro-Diels–Alder sequence which gave substantially higher yields than the known pyrolysis method, and was readily scalable. Overall pure sublimed H2O@C60 (78% H2O incorporation) was synthesised in 15% yield from C60. D2O@C60 uncontaminated with HOD@C60 was made in similar yield. The same synthetic route has been successfully applied to the formation of H2@C60, the large size of the orifice of compound 2 enabling a 60% incorporation of H2 at 120 °C and 120 atm. These advances will lead to a greatly improved availability of the important molecular endofullerenes H2O@C60 and H2@C60, and their isotopologues.
We thank the EPSRC (EP/I029451/1) and ERDF (Interreg-IVB, MEET project) for funding, and acknowledge the use of the IRIDIS High Performance Computing Facility and associated support services at the University of Southampton. We wish to thank Prof. Koichi Komatsu, Prof. Yasujiro Murata, Prof. Nick Turro, Dr Lei Xuegong and Dr YongJun Li for sharing their detailed experimental procedures.
Notes and references
- K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238–240 CrossRef CAS PubMed.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613–616 CrossRef CAS PubMed.
- M. H. Levitt, Philos. Trans. R. Soc., A, 2013, 371, 20120429 CrossRef PubMed.
- (a) A. J. Horsewill, K. Goh, S. Rols, J. Ollivier, M. R. Johnson, M. H. Levitt, M. Carravetta, S. Mamone, Y. Murata, J. Y.-C. Chen, J. A. Johnson, X. Lei and N. J. Turro, Philos. Trans. R. Soc., A, 2013, 371, 20110627 CrossRef CAS PubMed; (b) A. J. Horsewill, K. S. Panesar, S. Rols, M. R. Johnson, Y. Murata, K. Komatsu, S. Mamone, A. Danquigny, F. Cuda, S. Maltsev, M. C. Grossel, M. Carravetta and M. H. Levitt, Phys. Rev. Lett., 2009, 102, 013001 CrossRef CAS.
- C. Beduz, M. Carravetta, J. Y.-C. Chen, M. Concistré, M. Denning, M. Frunzi, A. J. Horsewill, O. G. Johannessen, R. Lawler, X. Lei, M. H. Levitt, Y. Li, S. Mamone, Y. Murata, U. Nagel, T. Nishida, J. Ollivier, S. Rols, T. Rõõm, R. Sarkar, N. J. Turro and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12894–12898 CrossRef CAS PubMed.
- (a) S. Mamone, M. Ge, D. Hüvonen, U. Nagel, A. Danquigny, F. Cuda, M. C. Grossel, Y. Murata, K. Komatsu, M. H. Levitt, T. Rõõm and M. Carravetta, J. Chem. Phys., 2009, 130, 081103 CrossRef CAS PubMed; (b) M. Ge, U. Nagel, D. Hüvonen, T. Rõõm, S. Mamone, M. H. Levitt, M. Carravetta, Y. Murata, K. Komatsu, X. Lei and N. J. Turro, J. Chem. Phys., 2011, 135, 114511 CrossRef PubMed; (c) T. Rõõm, L. Peedu, M. Ge, D. Hüvonen, U. Nagel, S. Ye, M. Xu, Z. Bačić, S. Mamone, M. H. Levitt, M. Carravetta, J. Y.-C. Chen, X. Lei, N. J. Turro, Y. Murata and K. Komatsu, Philos. Trans. R. Soc., A, 2013, 371, 20110631 CrossRef PubMed.
- S. Mamone, M. Concistrè, E. Carignani, B. Meier, A. Krachmalnicoff, O. G. Johannessen, X. Lei, Y. Li, M. Denning, M. Carravetta, K. Goh, A. J. Horsewill, R. J. Whitby and M. H. Levitt, J. Chem. Phys., 2014, 140, 194306 CrossRef PubMed.
- (a) M. Frunzi, S. Jockusch, J. Y.-C. Chen, R. M. K. Calderon, X. Lei, Y. Murata, K. Komatsu, D. M. Guldi, R. G. Lawler and N. J. Turro, J. Am. Chem. Soc., 2011, 133, 14232–14235 CrossRef CAS PubMed; (b) Y. Li, X. Lei, S. Jockusch, J. Y.-C. Chen, M. Frunzi, J. A. Johnson, R. G. Lawler, Y. Murata, M. Murata, K. Komatsu and N. J. Turro, J. Am. Chem. Soc., 2010, 132, 4042–4043 CrossRef CAS PubMed.
- (a) C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS; (b) R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- J. C. Hummelen, M. Prato and F. Wudl, J. Am. Chem. Soc., 1995, 117, 7003–7004 CrossRef CAS.
- (a) S. Iwamatsu, T. Uozaki, K. Kobayashi, S. Re, S. Nagase and S. Murata, J. Am. Chem. Soc., 2004, 126, 2668–2669 CrossRef CAS PubMed; (b) Q. Zhang, Z. Jia, S. Liu, G. Zhang, Z. Xiao, D. Yang, L. Gan, Z. Wang and Y. Li, Org. Lett., 2009, 11, 2772–2774 CrossRef CAS PubMed.
- J. C. Rasaiah, S. Garde and G. Hummer, Annu. Rev. Phys. Chem., 2008, 59, 713–740 CrossRef CAS PubMed.
- T. Futagoishi, M. Murata, A. Wakamiya, T. Sasamori and Y. Murata, Org. Lett., 2013, 15, 2750–2753 CrossRef CAS PubMed.
- (a) Q. Zhang, T. Pankewitz, S. Liu, W. Klopper and L. Gan, Angew. Chem., Int. Ed., 2010, 49, 9935–9938 CrossRef CAS PubMed; (b) Y. Guo, J. Yan and N. M. Khashab, ChemPhysChem, 2012, 13, 751–755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and details of calculations. See DOI: 10.1039/c4cc06198e |
| This journal is © The Royal Society of Chemistry 2014 |