
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bypassing a highly unstable frustrated Lewis pair: dihydrogen cleavage by a thermally robust silylium–phosphine adduct†
Thomas J.
Herrington
,
Bryan J.
Ward
,
Laurence R.
Doyle
,
Joe
McDermott
,
Andrew J. P.
White
,
Patricia A.
Hunt
* and
Andrew E.
Ashley
*
Department of Chemistry, Imperial College London, Exhibition Road, London SW7 2AZ, U.K. E-mail: a.ashley@imperial.ac.uk; Web: http://www3.imperial.ac.uk/people/a.ashley
First published on 26th August 2014
Abstract
The thermally robust silylium complex [iPr3Si–PtBu3]+[B(C6F5)4]− (1) activates H2/D2 at 90 °C (PhCl); no evidence for dissociation into the separated Lewis pair is found. DFT calculations show H2 cleavage proceeds via Si–P bond elongation to form an encounter complex directly from the adduct, thus avoiding the non-isolable iPr3Si+–PtBu3 FLP.
Since their discovery in 2006, ‘frustrated Lewis pairs’ (FLPs) have continued to provide a fresh and novel approach to the field of bond activation chemistry.1 FLPs can be defined as combinations of a Lewis acid and base which, by dint of steric hindrance, are unable to datively bind in the classical manner, leading to unquenched reactivity capable of activating small molecules. One of the most exciting properties of FLPs is their ability to induce heterolytic cleavage of H2 into protic and hydridic components (H+/H−),2 which can subsequently be transferred to reducible substrates either stoichiometrically (e.g. carbonyls, CO2)3 or catalytically (e.g. imines, silyl enol ethers, alkenes).4 By far the most commonly used Lewis acids are organometallics of the Group 13 elements (e.g. R3E; E = B or Al, R = electron-deficient organyl), among which B(C6F5)3 remains prevalent. Silylium ions (R3Si+) are both isoelectronic and isolobal with this class of compounds and, because of their potent electrophilicity,5 can demonstrate similar FLP reactivity when employed as the Lewis acid partner. However, there are few reports of stable R3Si+ cations, which is attributed to the difficulties in stabilising the diffuse 3p Si valence orbital either through hyper- or π-conjugation.6 This property explains, in part, their voracious affinity for nucleophiles; even traditionally ‘inert’ arene solvents7 and weakly coordinating anions (WCAs)8 can demonstrate interactions with the silicon centre. The work of Ozerov et al. reflects this extreme appetite for σ and π donors, in which decomposition of the robust [B(C6F5)4]− WCA was observed when heated in the presence of Et3Si+,9 presumably via [C6F5]− abstraction or C–F activation.10 Such indiscriminate reactivity may be suppressed via incorporation of intramolecular donors which moderate the potent electrophilicity while preserving sufficient reactivity for catalysis. In this respect, Oestreich and co-workers used the ferrocene-stabilised silylium ion [(C5H5)Fe(C5H4SitBuMe)]+ to selectively and catalytically hydrosilylate various ketones to the corresponding alkyl/silyl ethers (RR′CH–O–SiR3; R/R′ = alkyl);11 by contrast the fully reduced alkanes R–CH2–R′12 can be isolated when the ‘untamed’ ‘[R3Si]+’ is used as catalyst. Steric protection of the Si centre has also been adopted to prevent undesired silylation of aromatic solvents, as documented by Müller et al. Here, bulky triarylsilylium compounds (Ar3Si+; Ar = C6H6−xMex, x = 3–5), in conjunction with phosphine (R3P; R = alkyl, aryl) or silylene bases,13 are shown to generate FLPs that engage in H2 cleavage. Nonetheless, eventual solution-phase decomposition of these FLPs under ambient conditions was attributed to the long-term instability of the Ar3Si+ ions, which leads to protonated arene products.14
Herein we report the synthesis and characterisation of a classical donor–acceptor complex between tBu3P and the less sterically encumbered, highly reactive, silylium ion iPr3Si+. We show that the use of a Lewis adduct considerably stabilises the R3Si+ moiety, in comparison with the previously studied Ar3Si+ species. Furthermore, this species is shown to heterolytically cleave H2, the mechanism of which avoids the formation of a presumably highly unstable iPr3Si+–PtBu3 separated Lewis pair.
Treatment of iPr3SiH with [Ph3C][B(C6F5)4] (Bartlett–Condon–Schneider hydride transfer)15 in chlorobenzene afforded solutions of [iPr3Si·ClPh]+, as previously described.16 Subsequent in situ reaction with tBu3P furnished [iPr3Si–PtBu3]+[B(C6F5)4]− (1), upon precipitation with hexanes and recrystallisation from PhCl, in excellent yield (Scheme 1). 1 has been characterised by 1H, 13C, 29Si and 31P NMR spectroscopy, high resolution MS (ES+), and elemental analysis (see ESI†).
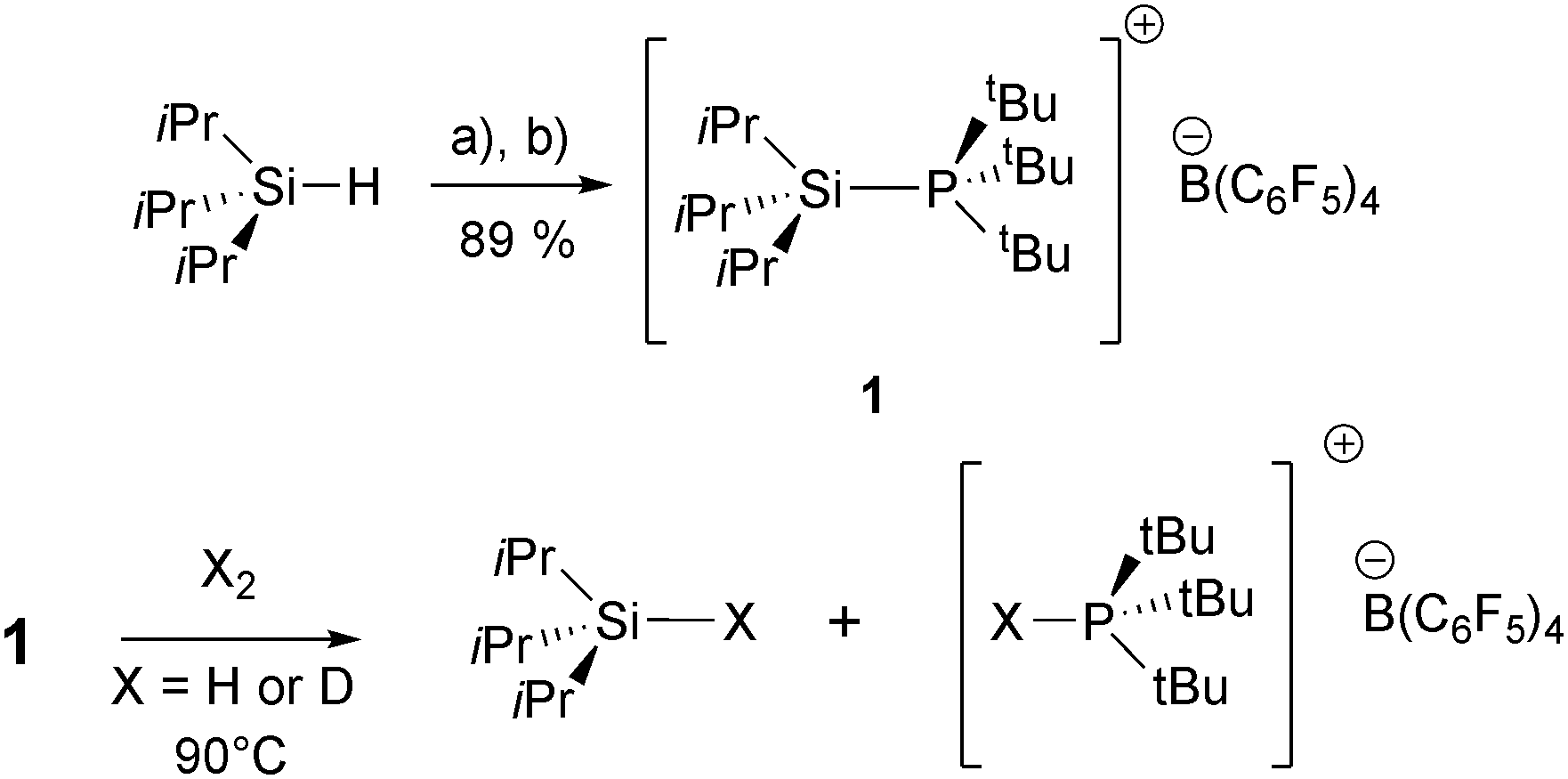 | ||
| Scheme 1 Synthesis of 1 and subsequent reactivity with H2/D2 (C6D5Cl or C6H5Cl solution, 0.096 M; H2 and D2 experiments respectively, 4 bar). Reagents and conditions: (a) (i) [Ph3C][B(C6F5)4], PhCl, RT, –Ph3CH; (b) tBu3P added in situ. | ||
Slow cooling of a PhF solution of 1 to −25 °C produced large colourless plates which were suitable for single crystal X-ray diffraction,‡ and the solid state structure is shown in Fig. 1.
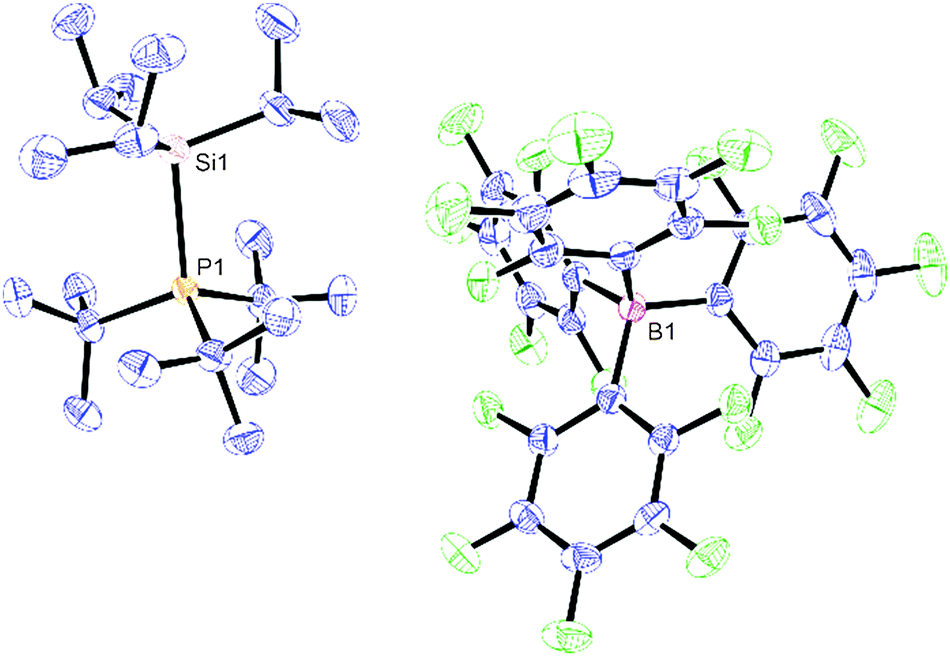 | ||
| Fig. 1 ORTEP plot of the X-ray structure of 1. C atoms blue, P atom orange, B atom pink, F atoms green and Si atom light brown. H atoms have been removed for clarity, and thermal ellipsoids are shown at 50% probability; Si1–P1 = 2.4843(5) Å. | ||
The [iPr3Si–PtBu3]+ fragment in 1 is derived from a donor–acceptor interaction between the iPr3Si+ moiety and tBu3P, as exemplified by the pyrimidalisation about the Si atom (0.5765(11) Å deviation from the plane of the three C atoms in the C3Si unit). The C–Si–C bond angles, which range between 110.51(8) and 111.86(9)°, are much closer to the idealised tetrahedral angle (109.5°) than those found in iPr3Si(CHB11H5Cl6) (117.3°),8 which possesses significant silylium character and hence approaches a trigonal geometry. The [B(C6F5)4]− anions are well separated from the cations, with no close Si to F contacts, and hence are non-coordinating. However, the Si–P bond distance is rather long (2.4843(5) Å; within the top 2% of those reported in the CSD), and may be compared to [PhMe2Si–PtBu3]+[HB(C6F5)3]− (Si–P = 2.376(2) Å), which has been prepared from reaction of PhMe2SiH and the FLP system tBu3P/B(C6F5)3.17 This increased distance may be attributed to the higher degree of steric strain due to crowding between the organic groups along the Si–P axis.
PhCl solutions of 1 proved to be stable for at least several months at room temperature, and these can be heated at 90 °C for 24 h without evidence of decomposition. 1H and 13C NMR spectra (C6D5Cl; 298 K) of 1 are commensurate with the solid state structure, and the upfield 29Si resonance (δ = 43.1 ppm, 1JSiP = 23 Hz) reveals the Si–P bond to be intact in solution. However, the coupling constant is noticeably smaller than those reported for [(C6Me5)3Si–PEt3]+[B(C6F5)4]− or zwitterionic [Mes2(SiPh2H)P+CH2CH2B−H(C6F5)2] (1JSiP = 41.2 and 48.5 Hz, respectively),14,18 and the 31P NMR resonance (δ = 57.3 ppm, 298 K) is close to that observed for free tBu3P (δ = 62.0 ppm, 298 K); collectively these data would suggest a weak Si–P bond.
In contrast to the results obtained using the silylium FLPs, (C6Me5)3Si+/PR3,14 admission of H2 to 1 (4 bar, C6D5Cl solvent) at room temperature led to no discernible reaction. However, heating these solutions to 90 °C led to complete consumption of the adduct (8 hours; Scheme 1), concomitant with formation of iPr3Si-H (1H NMR, δ = 3.43 ppm) and phosphonium borate [tBu3P-H]+[B(C6F5)4]− (1H NMR, δ = 4.17 ppm, 1JHP = 430 Hz; 31P {1H} NMR, δ = 60.3 ppm) in high conversion (90–94%, four runs).19 Conducting these experiments under D2 (C6H5Cl solvent) gave the deuterated products iPr3Si-D and [tBu3P-D]+, as shown by 2H, 31P and 29Si NMR spectroscopy (see ESI† for details);20 this conclusively shows that H2/D2 is the source of H/D atoms in the formally hydridic silane, and protic phosphonium ion.21 Upon reaction completion, 19F and 11B NMR spectra showed only resonances corresponding to the [B(C6F5)4]− counterion, demonstrating that silylium-mediated decomposition of the anion had not occurred. Since neither [H–B(C6F5)3]− nor B(C6F5)3 could be observed in solution, the possibility that H2 cleavage involves the known tBu3P/B(C6F5)3 FLP pathway17 may be discounted.
In order to investigate the possibility that 1 may dissociate in PhCl solution to generate tBu3P and solvated silylium ion, [iPr3Si·ClPh]+, a variable temperature (VT) 31P NMR experiment was conducted (Ph3P internal capillary reference; see Fig. S7 in ESI†). At low temperature the spectrum shows a very broad resonance (δ = 54.5 ppm, −40 °C) which moves progressively downfield (δ = 60.6 ppm, 100 °C), and markedly sharpens. However, it should be noted that the 31P NMR resonance for tBu3P also moves by ca. Δδ = 6 ppm over the same temperature range and the behaviour is likely due to a temperature-induced shift for both species. Furthermore, addition of tBu3P (1–10 eq., PhCl, 100 °C) to 1 produced 31P NMR spectra consisting only of their separate respective resonances; neither a discernible change in the lineshape nor chemical shift position of the adduct was observed. If rapid exchange between 1 and appreciable concentrations of dissociated products were indeed occurring, introduction of extraneous tBu3P would be expected to lead to a significant perturbation of the 31P NMR resonance of 1. Finally, we synthesised [iPr3Si·ClPh]+[B(C6F5)4]− in order to investigate its reactivity under the conditions of H2 activation, in the absence of added phosphine: at 90 °C this species degraded (40 min) via decomposition of the anion, producing B(C6F5)3, iPr3SiF (19F NMR δ = −185.0 ppm; 1JFSi = 298 Hz) and other unidentified products. This implies that, if dissociation of tBu3P from 1 were to occur and generate iPr3Si+ or (more likely) [iPr3Si·ClPh]+, the rate at which this cation reacts with [B(C6F5)4]− would greatly exceed the observed rate of H2 activation.
In order to determine whether thermally-induced dissociation of 1 (and hence a typical FLP-mediated mechanism) was responsible for H2 heterolysis, we examined this system further using DFT calculations. The results of our computational calculations (M06-2X/6-311+G(d,p) level of theory; see ESI†) which took into account secondary (conductor-like polarizable continuum model; C-PCM) solvent interaction, are presented in Scheme 2 (and Table S1, ESI†). Various conformations of 1 were identified (Table S2, ESI†), which are separated in free energy by only ca. 13–30 kJ mol−1; this lends support to the solution-phase VT 31P NMR data whereby the dynamic lineshape observed for 1 at low temperatures can be explained through interconversion between conformers. In total, two intermediates (A and B), and a single transition state for their interconversion (TSAB) were located along the reaction coordinate for H2 activation, starting from the lowest energy conformer of 1.
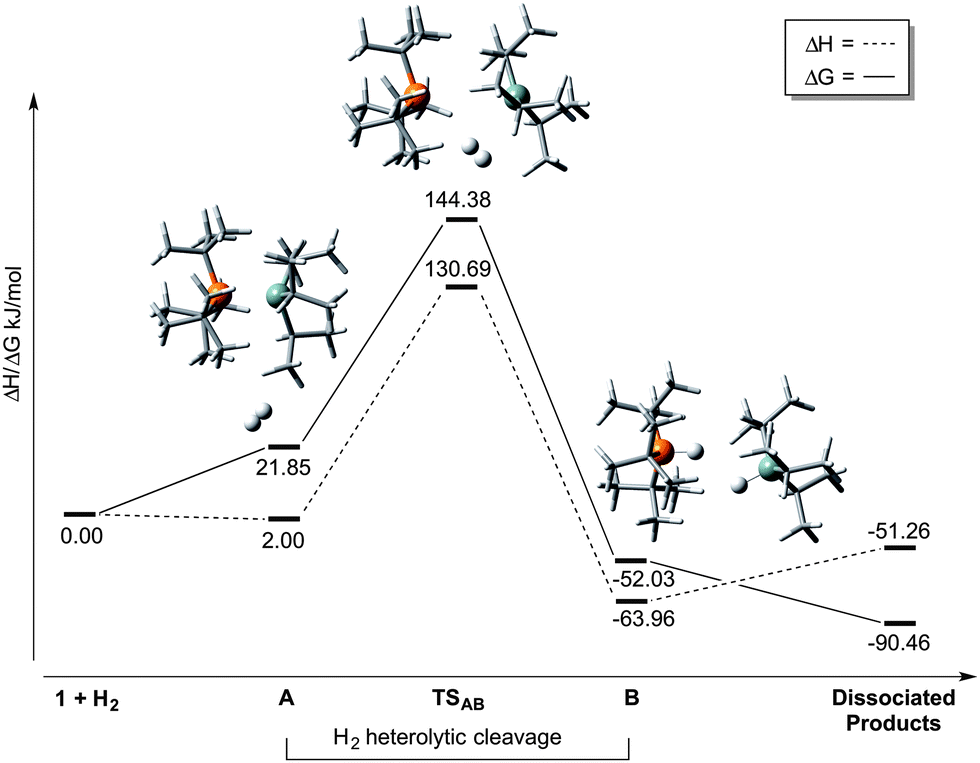 | ||
| Scheme 2 Solvent-phase enthalpy (ΔH) and Gibbs free energy (ΔG) profile for H2 activation mediated by 1 in PhCl solution, showing intermediates and transition states along the reaction coordinate (T = 363 K). P atom orange and Si atom pale green. | ||
On progressing to the transition state for H2 activation, TSAB, the Si⋯P distance lengthens considerably (Table 1), to an extent that is greater than the sum of the van der Waals radii of the elements (3.90 Å).22 The incipient cavity accompanying this bond elongation permits entry of a molecule of H2 while attractive electrostatic P⋯Si and secondary van der Waals interactions between iPr3Si and tBu3P fragments lead to binding beyond covalent distances, thus retarding tBu3P dissociation. This factor explains the experimental observation that the rate of H2 activation outcompetes decomposition, (which would be anticipated to be the faster process if dissociation to a true FLP were to occur at these elevated temperatures). A range of van der Waals interactions give rise to QTAIM bond critical points (BCP) between C–H⋯H–C and C–H⋯P (Tables S3 and S4, ESI†) which appear to hold the fragments in place, fulfilling a similar role to those of the C–H⋯F H-bonds reported for the frustrated encounter complex [tBu3P]⋯[B(C6F5)3] (the precursor species to H2 cleavage by that FLP system).23 It is notable that the B⋯P distance in the latter (4.20 Å) compares well with the Si⋯P length in TSAB, yet both are appreciably shorter than that those calculated for the ‘encounter complexes’ in Müller's (C6Me5)3Si+/PR3 systems (R = alkyl, aryl; range 4.45–5.73 Å); this likely reflects the much greater steric bulk of the Ar3Si+ fragment in these ‘true’ FLPs.14 Heterolytic cleavage of H2 subsequently proceeds to give [tBu3P-H]+ and H-SiiPr3 as a dihydrogen bonded intermediate (B), after which dissociation to form the free products is strongly entropically driven. The TS molecular orbitals exhibit H2 contributions (Fig. S19, ESI†) while BCPs from H2 to both P and Si are obtained (Table S5, ESI†). Moreover, the early TS exhibits nascent NBO P → H2(σ*) and H2(σ) → Si electron transfer, approximately equal to 8 and 34 kJ mol−1 respectively, which are expected to increase as the reaction proceeds. Heterolysis of H2 in this manner is consistent with the electron transfer model,24 with no observable deviation in H–H distance between that in TSAB and free H2, denoting an early transition state.
Formation of TSAB is both enthalpically unfavourable owing to the weakening of the Si–P interaction, and entropically unfavourable due to the increased ordering as a result of H2 coordination (Table S1, ESI†). The substantial energy barrier (122.53 kJ mol−1; A→TSAB) associated with H2 activation is testament to the strong Si–P dative bond in 1 and explains the elevated temperatures required experimentally to achieve bond elongation and access the encounter complex. Comparable rate-determining steps have been observed elsewhere in the literature; Lammertsma et al. report that the experimentally observed insertion of CO2 into dimeric P/Al-based Lewis pairs proceeds at room temperature, despite having computed a significant energy barrier (ca. 140 kJ mol−1).25
Whilst a number of Lewis pairs have been documented that exhibit classical/frustrated borderline reactivity with H2 or alkynes,26 spectroscopic evidence for the existence of the dissociated constituents has always been demonstrated, due to the stability of the Lewis acid as an independent entity. In our particular example, however, we have shown that the critical development of an encounter complex prior to H2 activation can be achieved directly from the Lewis pair adduct via simple bond elongation and weakening, obviating the need for the formation of a true FLP; this is especially important when the Lewis acid partner (in our case iPr3Si+) is too reactive to isolate in the free form.
In conclusion, we have shown that a classical donor–acceptor adduct incorporating a highly electrophilic trialkylsilylium ion can activate H2; this reaction is thus not rigidly confined to truly separated R3Si+/base FLP combinations. Indeed, the formation of a stable adduct incorporating such species may provide a general strategy towards the protection of highly reactive Lewis acids/bases in Lewis pair systems, without excluding such systems from participating in characteristic FLP-type chemistry. We are currently exploring the small molecule reactivity of other [R3Si–(base)]+ adducts, in addition to investigating their potential use in the catalytic hydrogenation of unsaturated substrates.
The authors wish to acknowledge the Royal Society for a University Research Fellowship (AEA), the EPSRC for studentship funding (TJH and BJW), and Dr Richard Matthews for his assistance with the QTAIM analysis.
Notes and references
- (a) G. Erker and D. W. Stephan, Frustrated Lewis Pairs I: Uncovering and Understanding, Top. Curr. Chem., Springer GmbH, Berlin, 2013, pp. 1–350 Search PubMed; (b) G. Erker and D. W. Stephan, Frustrated Lewis Pairs II: Expanding The Scope, Top. Curr. Chem., Springer GmbH, Berlin, 2013, pp. 1–350 Search PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- (a) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839 CrossRef CAS PubMed; (b) G. Menard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef CAS PubMed; (c) V. Sumerin, F. Schulz, M. Nieger, M. Leskela, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS PubMed.
- (a) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS PubMed; (b) L. Greb, S. Tussing, B. Schirmer, P. Ona-Burgos, K. Kaupmees, M. Lokov, I. Leito, S. Grimme and J. Paradies, Chem. Sci., 2013, 4, 2788 RSC.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 325 CrossRef CAS.
- A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 4526 CrossRef CAS PubMed.
- J. B. Lambert, S. H. Zhang, C. L. Stern and J. C. Huffman, Science, 1993, 260, 1917 CAS.
- Z. W. Xie, J. Manning, R. W. Reed, R. Mathur, P. D. W. Boyd, A. Benesi and C. A. Reed, J. Am. Chem. Soc., 1996, 118, 2922 CrossRef CAS.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed.
- M. F. Ibad, P. Langer, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2011, 133, 21016 CrossRef CAS PubMed.
- K. Muether and M. Oestreich, Chem. Commun., 2011, 47, 334 RSC.
- M. Kira, T. Hino and H. Sakurai, Chem. Lett., 1992, 555 CrossRef CAS.
- (a) A. Schaefer, M. Reissmann, A. Schaefer, W. Saak, D. Haase and T. Mueller, Angew. Chem., Int. Ed., 2011, 50, 12636 CrossRef CAS PubMed; (b) A. Schäfer, M. Reißmann, A. Schäfer, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2014, 20, 9381 CrossRef PubMed.
- M. Reißmann, A. Schäfer, S. Jung and T. Müller, Organometallics, 2013, 32, 6736 CrossRef.
- P. D. Bartlett, F. E. Condon and A. Schneider, J. Am. Chem. Soc., 1944, 66, 1531 CrossRef CAS.
- A. Schaefer, W. Saak, D. Haase and T. Mueller, Angew. Chem., Int. Ed., 2012, 51, 2981 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed.
- W. Nie, H. F. T. Klare, M. Oestreich, R. Froehlich, G. Kehr and G. Erker, Z. Naturforsch., B: Chem. Sci., 2012, 67, 987 CrossRef CAS.
-
1 proved to be highly reactive to even trace amounts of H2O. Accordingly, the synthesis and solution-phase studies required meticulous drying of solvents, conducting reactions in Teflon® vials, and performing NMR experiments in Teflon® inserts. Reactions in dilute solution, such as those under H2, invariably become exposed to trace amounts of H2O, leading to [tBu3PH]+[B(C6F5)4]− and (iPr3Si)2O (2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The yield for H2 conversion is most reliably calculated via relative integration of 1H NMR signals for the iPr3Si-H resonance against that of tBu3P-H (1:1 from H2). Accordingly, hydrolysis produces twice the amount of phosphonium salt than the reaction with H2, and accounts for the slightly sub-quantitative yields (90–94%). In support of this conclusion, 2H NMR spectroscopy of the D2 cleavage reaction shows a 1:1 tBu3P-D:D-SiiPr3 ratio, since adventitious moisture is introduced as H2O, and not D2O (see Fig. S8 in ESI†).
:1). The yield for H2 conversion is most reliably calculated via relative integration of 1H NMR signals for the iPr3Si-H resonance against that of tBu3P-H (1:1 from H2). Accordingly, hydrolysis produces twice the amount of phosphonium salt than the reaction with H2, and accounts for the slightly sub-quantitative yields (90–94%). In support of this conclusion, 2H NMR spectroscopy of the D2 cleavage reaction shows a 1:1 tBu3P-D:D-SiiPr3 ratio, since adventitious moisture is introduced as H2O, and not D2O (see Fig. S8 in ESI†). - A full kinetic study by 1H NMR was hampered by iPr3SiH interaction with 1, which masks the actual amount of silane produced until reaction completion (i.e. complete conversion of 1). This phenomenon has been noted for R3Si(μ-H)SiR3+ species,28 wherein the silane proton is rendered invisible to 1H NMR spectroscopy. It is plausible that a similar bridging silane interaction is also operational here.
- Heating solutions of independently prepared [tBu3P-H]+[B(C6F5)4]− with iPr3SiH (PhCl, 90 °C, 60 h) led to no reaction, demonstrating that the H2 cleavage reaction is irreversible. Furthermore, conducting the same experiments under D2 did not produce any HD, which would be expected to form due to H/D scrambling.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435 CrossRef CAS PubMed.
- T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425 CrossRef CAS PubMed.
- F. Bertini, F. Hoffmann, C. Appelt, W. Uhl, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Organometallics, 2013, 32, 6764 CrossRef CAS.
- (a) S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476 CrossRef CAS PubMed; (b) S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842 RSC; (c) C. Jiang and D. W. Stephan, Dalton Trans., 2013, 42, 630 RSC; (d) T. Voss, T. Mahdi, E. Otten, R. Frohlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367 CrossRef CAS; (e) S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884 RSC; (f) S. J. Geier, A. L. Gille, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2009, 48, 10466 CrossRef CAS PubMed; (g) L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164 CrossRef CAS PubMed; (h) E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2012, 18, 16938 CrossRef CAS PubMed; (i) C. Jiang, O. Blacque, T. Fox and H. Berke, Organometallics, 2011, 30, 2117 CrossRef CAS; (j) M. Ullrich, A. J. Lough and D. W. Stephan, Organometallics, 2010, 29, 3647 CrossRef CAS; (k) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594 CrossRef CAS.
- SHELXTL, SHELX-2013, Bruker AXS, Madison WI, http://shelx.uni-ac.gwdg.de/SHELX/index.php Search PubMed.
- (a) M. Nava and C. A. Reed, Organometallics, 2011, 30, 4798 CrossRef CAS PubMed; (b) S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section including computational details, characterisation data and copies of the NMR spectra compounds and reactions. CCDC 970728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc05905k |
‡ Crystallographic data for 1: C45H48BF20PSi, M = 1038.70, triclinic, a = 11.6772(5) Å, b = 12.5736(4) Å, c = 17.0286(6) Å, α = 79.442(3)°, β = 75.102(3)°, γ = 77.292(3)°, U = 2335.61(15) Å3, T = 173 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 2, ρcalcd = 1.477 g cm−3, μ(CuKα) = 1.792 mm−1, colourless blocks, Agilent Xcalibur PX Ultra A diffractometer; 8923 independent measured reflections (Rint = 0.0180), F2 refinement,27R1(obs) = 0.0354, wR2(all) = 0.0948, 7708 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 147°], 613 parameters. CCDC 970728. (no. 2), Z = 2, ρcalcd = 1.477 g cm−3, μ(CuKα) = 1.792 mm−1, colourless blocks, Agilent Xcalibur PX Ultra A diffractometer; 8923 independent measured reflections (Rint = 0.0180), F2 refinement,27R1(obs) = 0.0354, wR2(all) = 0.0948, 7708 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 147°], 613 parameters. CCDC 970728. |
| This journal is © The Royal Society of Chemistry 2014 |