
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disentangling scalar coupling patterns by real-time SERF NMR†
Nina
Gubensäk
,
Walter M. F.
Fabian
and
Klaus
Zangger
*
Institute of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: klaus.zangger@uni-graz.at
First published on 27th August 2014
Abstract
Scalar coupling constants and signal splitting patterns in NMR spectra contain a wealth of short-range structural information. The extraction of these parameters from 1H NMR spectra is often prohibited by simultaneous scalar coupling interactions with several other protons. Here we present a high-resolution NMR experiment where scalar coupling to only one selected signal is visible. All other couplings are removed from the spectrum. This real-time selectively refocused NMR experiment is achieved by spatially selective homonuclear broadband decoupling combined with selective refocusing during acquisition. It allows the unperturbed extraction of scalar coupling constants from the highly resolved acquisition dimension of NMR spectra.
NMR spectroscopy is one of the most frequently used techniques for the structural characterization of small to medium sized organic molecules and biomolecules. Because of their widespread occurrence, high natural abundance and high sensitivity, 1H nuclei are often used in this process. Structural information is obtained from the resonance frequencies and scalar coupling patterns. However, due to the limited chemical shift range of protons, the signals are often overlapped, rendering the extraction of structural information difficult or sometimes impossible. One way to increase the resolution of 1H NMR spectra is the use of pure-shift methods,1–7 which yield singlet only spectra, reminiscent of 13C NMR spectra. While these experiments provide highest resolution 1D proton NMR spectra, scalar coupling information, which is often key in analyzing chemical structures, is of course completely lost in such experiments. Several methods have been described in order to simplify spectra (increase their resolution) and still allow the extraction of scalar coupling information. Most commonly two-dimensional E-COSY type spectra,8 soft-COSY9 or the MUSIC experiment10 are employed, yielding multiplets where active and passive couplings are separated or 2D J-resolved experiments,2 separating the chemical shift in the direct dimension from J-coupling information in the indirect dimension. For complicated multiplets or larger molecules these spectra still often result in crowded multiplet patterns. One technique to significantly simplify scalar coupling patterns and therefore enable the extraction of individual couplings is the SERF (selective refocusing) experiment proposed by Fäcke and Berger.11 In the original version, two coupling partners are selectively excited and only their mutual scalar coupling evolves during t1, resulting in a two-dimensional J-resolved experiment showing the splitting between the two selected spins only. This method allows the unperturbed determination of scalar coupling constants but it would be impractical for a larger number of coupling constants, as n(n − 1) two-dimensional experiments are required to measure all coupling constants in a system of n coupled signals. Several extensions and improvements of one- and two-dimensional SERF spectra have been described and applied to several systems.12–15 Recently, the parallel acquisition of all SERF spectra connected to one selected peak has been described by Giraud and Merlet et al.16 For this experiment, a selective pulse is applied during a weak pulsed field gradient, resulting in the excitation of the whole spectrum, but different signals are selected in different slices of the sample tube. Subsequently, a spatially selective 2D SERF spectrum is recorded, where each signal stems from a different slice of the NMR sample. Coupling constants are again extracted from a 2D J-resolved spectrum. The concept of “parallel data acquisition”17,18 in different spatially separated regions is mainly used for in vivo NMR spectroscopy, but has found its way into liquid-state NMR through several experiments, most notable single-scan (ultra-fast) spectroscopy,19,20 but also for homonuclear broadband decoupling,7 relaxation-delay free data acquisition21,22 or diagonal-peak suppressed homonuclear 2D NMR.23 For the latter experiments and also the spatially-separated 2D SERF experiment, the gain in resolution comes at the price of a significantly reduced sensitivity since each signal originates from a narrow slice of the sample tube. Combined with the need to record 2D spectra for all these SERF experiments, long measurement times are usually required.
Here we present a real-time (single scan) experiment, which allows the recording of one-dimensional spectra showing all scalar couplings to one selected signal only. Signals not directly coupled to the selected spin are reduced to singlets. Limiting the scalar coupling evolution during acquisition is achieved by spatially selective scalar coupling refocusing between individual FID data chunks using the pulse sequence presented in Fig. 1. After a 90° selective pulse applied during a weak pulsed field gradient (∼1–2 Gauss cm−1) the acquisition of the first FID data block is started. Between successive FID chunks scalar coupling is refocused by a combination of a hard and a spatially-selective 180° pulse, similar to slice-selective homonuclear broadband decoupling (also called the Zangger–Sterk or ZS method).1,5,7,24 However, scalar coupling to one selected proton is allowed to evolve by the two additional 90° pulses applied to the frequency of the desired coupling partner. The result is a 1D 1H spectrum, which shows only the active coupling to the selectively refocused signal and which does not require any special data processing. The individual data chunks are automatically added during the execution of the pulse sequence. The principle of this experiment is similar to 2D SERF spectra, but the scalar coupling information is obtained from the directly acquired spectrum rather than the indirect dimension of a J-resolved type experiment. The presented experiment should not be confused with selective 1D experiments, where only one signal remains in the spectrum. In such a 1D COSY or TOCSY there is still the whole coupling pattern present while the real-time SERF experiment reduces the coupling to only one selected partner. Besides the significant reduction in experimental time, another big advantage of real-time SERF is that the signal splitting is observed in the detection dimension, where high resolution is readily achieved by a large number of acquired data points. In contrast 2D experiments, which provide the scalar coupling information in the indirect dimension, need to be recorded with a large number of increments to achieve high resolution and this requires proportionally longer measurement times. The presented experiment is the only available technique which allows the restriction of scalar coupling to only one partner during acquisition and therefore the recording of scalar coupling limited 1D spectra.
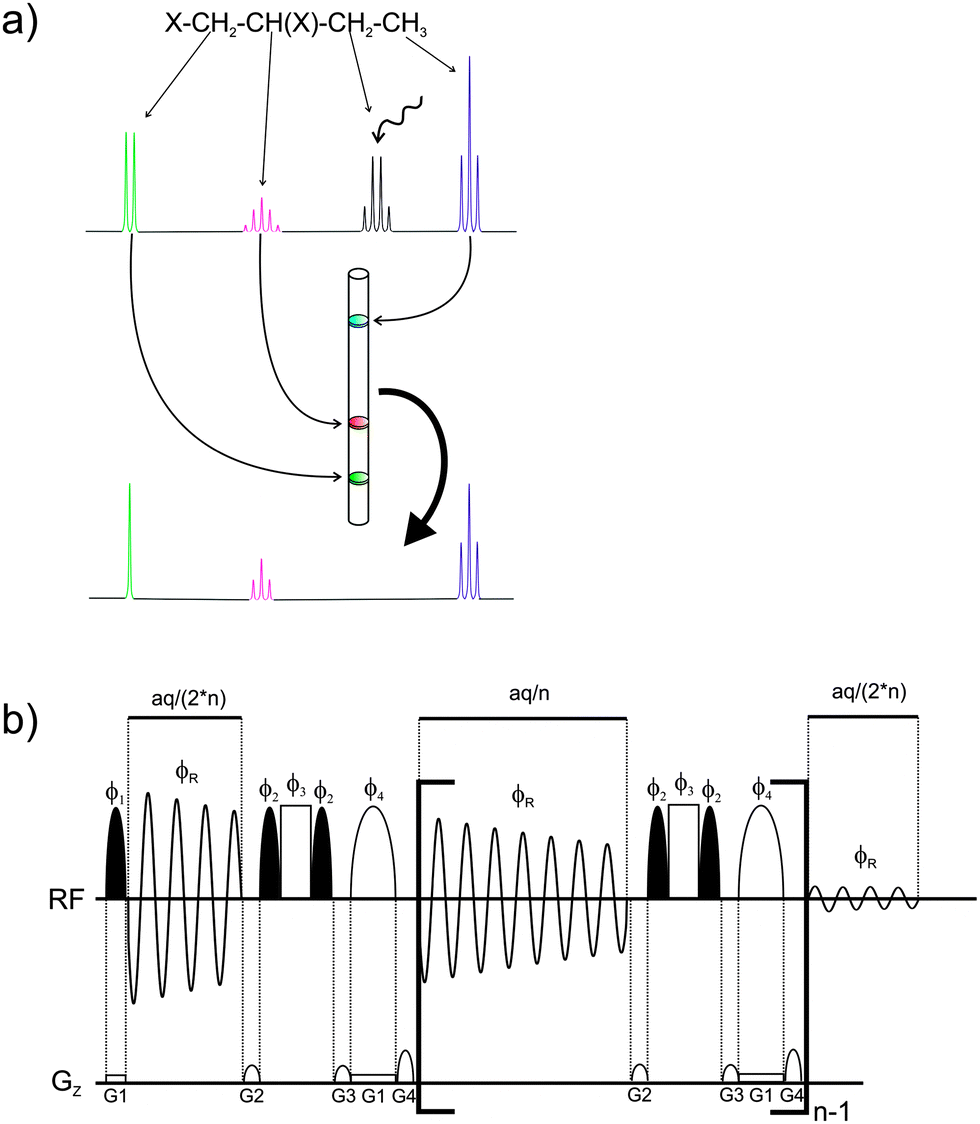 | ||
Fig. 1 A schematic representation of the real-time SERF NMR experiment is shown in (a): each signal is excited in a single slice of the sample tube and after frequency-selective refocusing of one chosen signal only scalar coupling to this signal is observable. The pulse-sequence of the 1D real-time SERF experiment is presented in (b). Black and white half-ellipsoids are selective 90° and 180° pulses, respectively. White rectangles are hard 180° pulses. The total acquisition time is aq and the pulse phases are ϕ1 = ϕR = x, −x, −x, x, y, −y, −y, y; ϕ2 = x, x, −x, −x; ϕ3 = x, −x; ϕ4 = −x, x. The relative gradient strengths are G2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G3:G4 = −1:1:2. The strength of G1, which is the gradient for slice-selective excitation, depends on the spectral width. It is typically ∼1–2 G cm−1. :G3:G4 = −1:1:2. The strength of G1, which is the gradient for slice-selective excitation, depends on the spectral width. It is typically ∼1–2 G cm−1. | ||
As a first example of the presented method, a series of real-time SERF spectra of n-propanol can be seen in Fig. 2. The selectively recoupled signal in each spectrum is indicated by the wavy arrows. The resulting spectra yield significantly simplified scalar coupling patterns, which show only the coupling to the selected resonance. Therefore, coupling constants can be extracted much more easily and accurately. As an example, the apparent quartet for the CH2– group at position 1, with a “coupling constant” of ∼6.6 Hz actually consists of a doublet (coupling to the OH proton) with J = 5.2 Hz and a triplet (coupling to CH2 at position 2) with J = 6.5 Hz. A similar situation is encountered for the H-2 signal, where an obvious sextet with J = 6.8 Hz is split into a quartet with J = 7.1 Hz and a triplet with J = 6.5 Hz. These spectra were recorded on a sample containing 48 mg n-propanol in 600 μl of DMSO-d6 by accumulating 256 scans (experimental time ∼12 min). The sensitivity of real-time SERF spectra is of course markedly reduced compared to regular 1D spectra as a result of spatially selective excitation. The loss in sensitivity depends mainly on the required spectral width and the selectivity of the decoupling pulse. For a 10 ppm range and a 10 ms Gaussian pulse the total signal intensity of the selectively refocused spectrum is about 3% of a regular 1D spectrum. However, some of this loss is regained by the partial removal of couplings and the narrower linewidths as a result of increased magnetic field homogeneity in a single slice as compared to the whole detection volume. Spectra obtained with only 1 scan on a sample of 8 mg propanol in 600 μl of DMSO-d6 are found in the ESI† to show that even without cryo probe technology, useful one-dimensional real-time SERF spectra can be obtained on low-concentration samples in a matter of seconds.
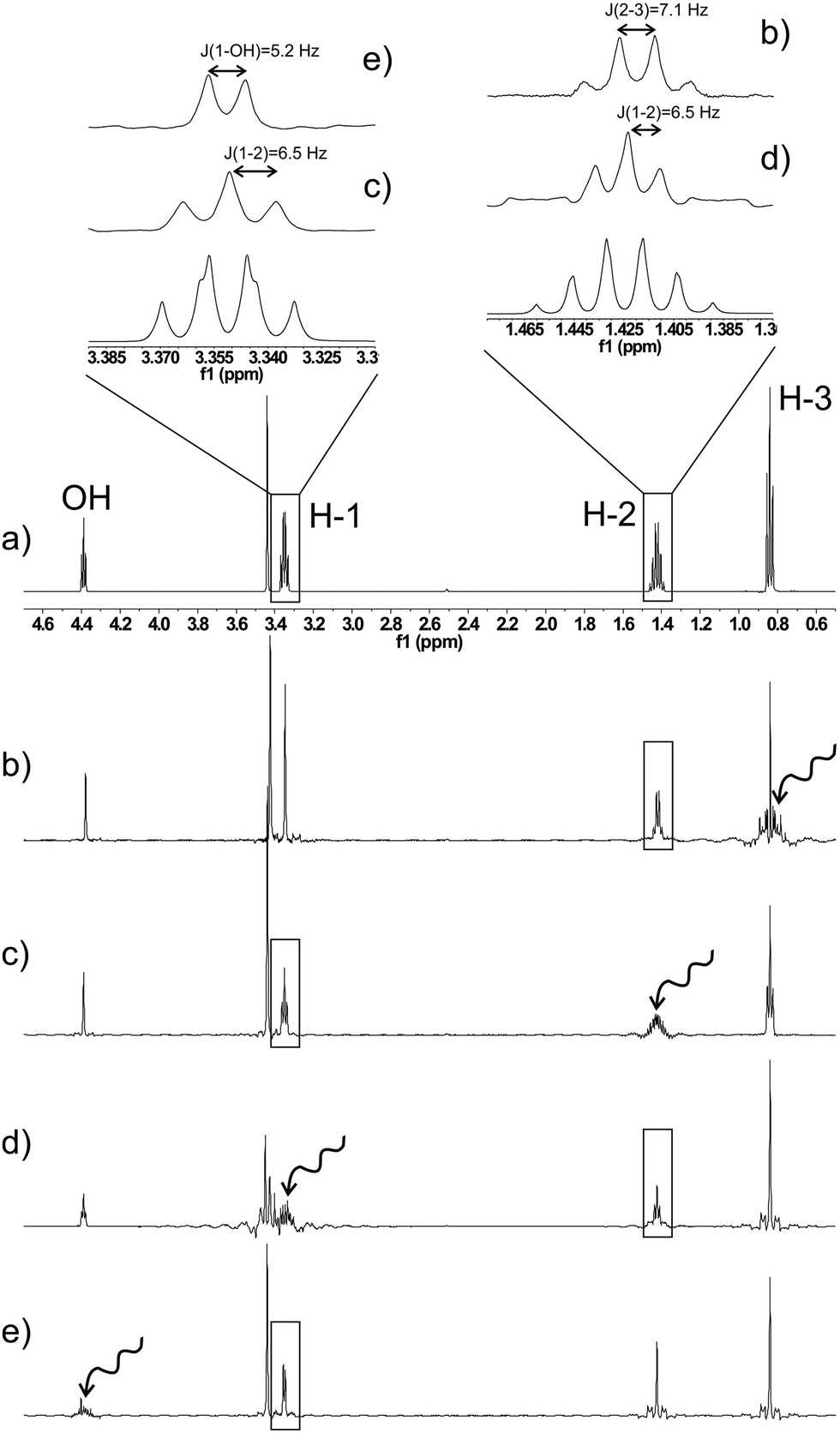 | ||
| Fig. 2 Regular one-dimensional 1H NMR spectrum (a) and real-time SERF spectra (b–e) of n-propanol. The signals which are selectively recoupled in the individual spectra are indicated by wavy arrows. The regions of the H-1 and H-2 signals are enlarged. The regular spectrum shows an apparent quartet for H-1 and a sextet for H-2. However, the SERF spectra reveal that both signals actually consist of components with coupling constants differing by 0.7 and 0.6 Hz, for H-1 and H-2, respectively. | ||
While in the case of n-propanol more accurate coupling constants can be obtained with SERF spectra, in other cases the appearance of the multiplet could render the extraction of J values completely impossible. The 1D proton spectrum of cyclo-phenylalanine-proline (cyclo-Phe-Pro, cFP) is an example (Fig. 3).
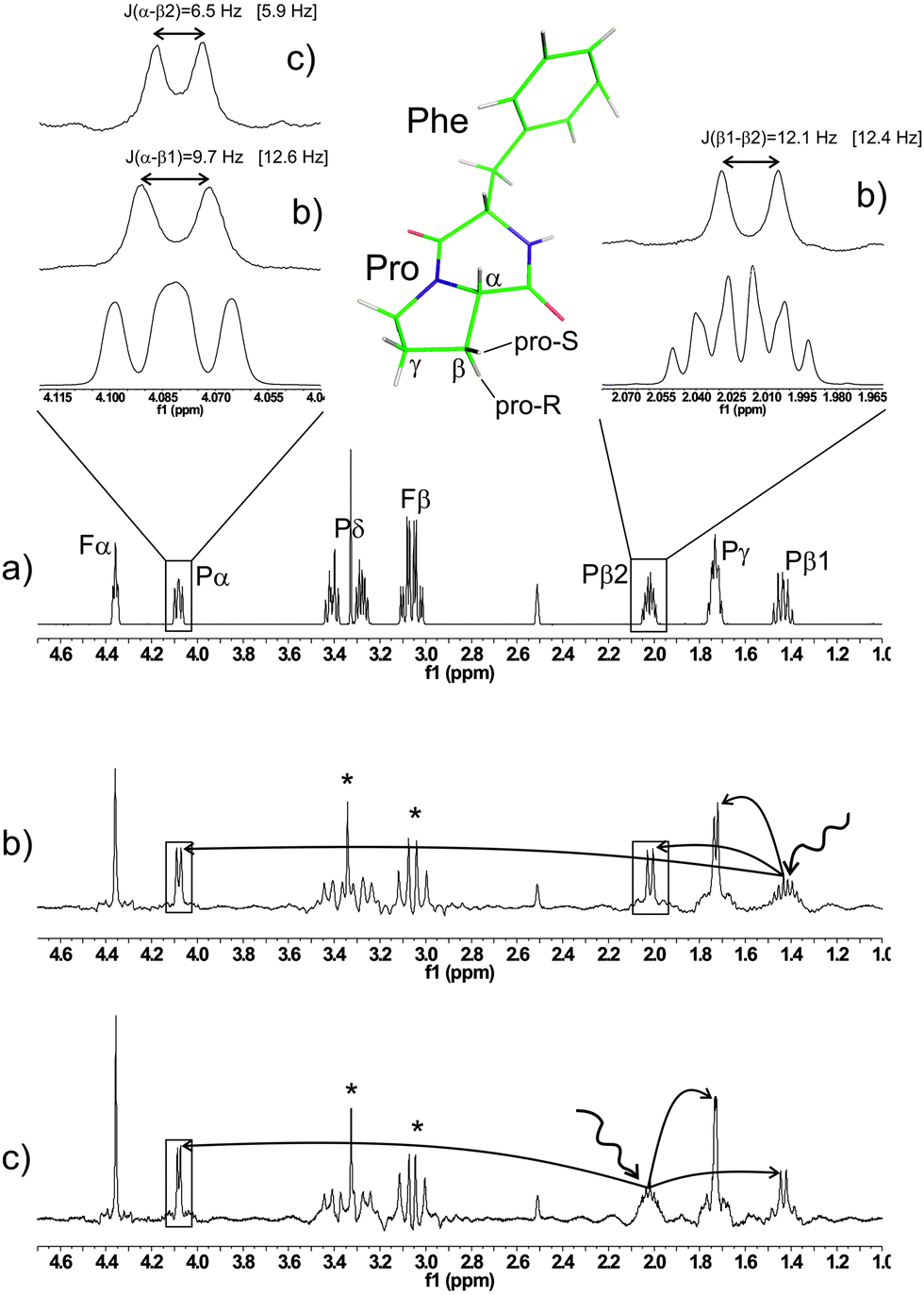 | ||
| Fig. 3 Scalar coupling constants involving the proline β-protons of cyclo-Phe-Pro cannot be obtained from a regular 1H spectrum (a), while they are all obtained from the real-time SERF spectra in (b) and (c). The proline β1 shows the larger 3J value to the α proton (9.7 Hz), whereas 3Jα-β2 = 6.5 Hz. This allows the prochiral assignment of β1 being the pro R and β2 the pro S nucleus. Calculated scalar coupling constants are given in square brackets. Artifacts resulting from strong coupling within the proline δ and phenylalanine β protons are indicated by asterisks. A density-functional optimized structure of cFP in DMSO solvent is shown as a stick model. | ||
All signals involving either the α or β protons are rather complex and accurate coupling constants cannot be obtained. The extreme simplification of 1D SERF spectra, shown in Fig. 3 delivers all J values as indicated. Proton β1 shows a larger coupling to H-α (J = 9.7 Hz) compared to β2 (J = 6.5 Hz), which indicates that β1 is the transoid pro-R site. This is corroborated by coupling constants obtained by density functional calculations on a geometry-optimized model of cFP in DMSO (see Fig. 3). The spectra shown in Fig. 3 were obtained using a 39 mg in 600 μl DMSO-d6 sample of cyclo-Phe-Pro and acquiring 256 scans, which amounts to a total acquisition time of ∼12 min for each 1D spectrum.
The concept of real-time SERF experiments can of course also be extended to two-dimensional NMR spectra. This might be necessary when the resolution of a one-dimensional decoupled SERF spectrum is not sufficient. To obtain an ω2 real-time SERF version of any kind of 2D spectrum, a 90° pulse right before acquisition needs to be replaced by a slice-selective 90° pulse and the acquisition scheme of Fig. 1 is then used during detection. One important feature of real-time SERF spectra is that the coupling constant is obtained from the signal splitting in the direct dimension (ω2 in 2D spectra), where the resolution can be set rather high without the need for long experimental times. In contrast, in regular SERF spectra the coupling constant is obtained from the indirect dimension, where high resolution goes hand in hand with long measurement times.
While the presented spectra yield the recoupled signals of one selected spin only, it would be possible to extend this concept to a two-dimensional COSY-type spectrum, where the chemical shift of the selected signal is spatially encoded in the indirect dimension using a second pulsed-field gradient direction (x or y). This would allow the scalar coupling constants of all signals to be obtained at the same time from the ω2-dimension, but at the cost of an additional huge sensitivity loss related to the double spatially selective (voxel-selective) excitation. Such an experiment would contain information similar to a spectrum described by Giraud and Merlet et al., a 2D combined J-edited and correlated experiment.25 However, it would show the splitting by scalar coupling in the high-resolution ω2, rather than in the indirect dimension.
In conclusion, real-time SERF spectra allow the extraction of scalar coupling constants to one selected signal, while reducing all other signals to singlets. This not only greatly simplifies the spectra, but in many cases would be the only way to extract individual coupling constants from 1D spectra. The selective recoupling during acquisition also allows a higher resolution for the reduced multiplets when compared to two-dimensional spectra where the resolution is given by the number of increments. Despite the significant signal reduction as a result of slice-selective excitation, coupling constants can be obtained from spectra acquired with only one scan on samples at millimolar concentrations.
Financial support to K.Z. from the Austrian Science Foundation (FWF) under project number P24742 is gratefully acknowledged. We also thank the interuniversity program in natural sciences, NAWI Graz, for financial support.
Notes and references
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS PubMed.
- W. P. Aue, J. Karhan and R. R. Ernst, J. Chem. Phys., 1976, 64, 4226–4227 CrossRef CAS PubMed.
- M. Foroozandeh, R. W. Adams, N. J. Meharry, D. Jeannerat, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2014, 53, 6990–6992 CrossRef CAS PubMed.
- A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS PubMed.
- N. H. Meyer and K. Zangger, Angew. Chem., Int. Ed., 2013, 52, 7143–7146 CrossRef CAS PubMed.
- N. H. Meyer and K. Zangger, ChemPhysChem, 2014, 15, 49–55 CrossRef CAS PubMed.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- C. Griesinger, O. W. Soerensen and R. R. Ernst, J. Am. Chem. Soc., 1985, 107, 6394–6396 CrossRef CAS.
- L. Emsley, P. Huber and G. Bodenhausen, Angew. Chem., Int. Ed., 1990, 29, 517–520 CrossRef PubMed.
- N. Müller, L. Di Bari and G. Bodenhausen, J. Magn. Reson., 1991, 94, 73–81 Search PubMed.
- T. Fäcke and S. Berger, J. Magn. Reson., Ser. A, 1995, 113, 114–116 CrossRef.
- B. Baishya, U. R. Prabhu and N. Suryaprakash, J. Phys. Chem., 2007, 111, 12403–12410 CrossRef CAS PubMed.
- M. E. Di Pietro, C. Aroulanda and D. Merlet, J. Magn. Reson., 2013, 234, 101–105 CrossRef CAS PubMed.
- D. Merlet, L. Beguin, J. Courtieu and N. Giraud, J. Magn. Reson., 2011, 209, 315–322 CrossRef CAS PubMed.
- J. M. Nuzillard, J. Magn. Reson., 2007, 187, 193–198 CrossRef CAS PubMed.
- N. Giraud, L. Beguin, J. Courtieu and D. Merlet, Angew. Chem., Int. Ed., 2010, 49, 3481–3484 CrossRef CAS PubMed.
- E. Kupce, R. Freeman and B. K. John, J. Am. Chem. Soc., 2006, 128, 9606–9607 CrossRef CAS PubMed.
- P. Nolis, M. Perez-Trujillo and T. Parella, Angew. Chem., Int. Ed., 2007, 46, 7495–7497 CrossRef CAS PubMed.
- L. Frydman, T. Scherf and A. Lupulescu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15858–15862 CrossRef CAS PubMed.
- A. Tal and L. Frydman, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 241–292 CrossRef CAS PubMed.
- P. Sakhaii, B. Haase, W. Bermel, R. Kerssebaum, G. E. Wagner and K. Zangger, J. Magn. Reson., 2013, 233, 92–95 CrossRef CAS PubMed.
- G. E. Wagner, P. Sakhaii, W. Bermel and K. Zangger, Chem. Commun., 2013, 49, 3155–3157 RSC.
- S. Glanzer, E. Schrank and K. Zangger, J. Magn. Reson., 2013, 323, 1–6 CrossRef PubMed.
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC.
- N. Giraud, D. Pitoux, J. M. Ouvrard and D. Merlet, Chemistry, 2013, 19, 12221–12224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, single-scan 1D SERF spectra and the Bruker format pulse sequence. See DOI: 10.1039/c4cc05892e |
| This journal is © The Royal Society of Chemistry 2014 |