
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bronze, silver and gold: functionalized group 11 organotin sulfide clusters†
Jens P.
Eußner
and
Stefanie
Dehnen
*
Philipps-Universität Marburg, Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Hans-Meerwein-Straße, D-35043 Marburg, Germany. E-mail: dehnen@chemie-uni-marburg.de; Fax: +49 6421 2825653; Tel: +49 6421 2825751
First published on 4th August 2014
Abstract
The synthesis, properties and reactivity of group 11 organotin sulfide clusters [(R1Sn)4(SnCl)2(MPPh3)2S8] (M = Cu, Ag), [(R3Sn)10Ag10S20], and [(R1,3Sn)2(AuPPh3)2S4] with covalently bound, carbonyl or hydrazine-terminated ligands R1 = CMe2CH2C(Me)O or R3 = CMe2CH2C(Me)NNH2 are reported.
The last few decades have afforded a large variety of ligand-protected chalcogenide clusters of group 11 metals.1 Ternary complexes and clusters were also reported,2 which often combined structural and physical properties of the binary components.3 Additionally, with regard to further reactivity, the introduction of functionalized phosphines was achieved.4 Consequently, the combination of functional organic ligand shells with finely tunable (ternary or multinary) inorganic cores, providing even more diversity, is thus of interest for diverse purposes, such as for optoelectronics or solar cell development.5 To address suitable functional materials, the synthesis and directed derivatization of clusters with covalently attached organic groups have currently been investigated in detail for polyoxometalates6 as well as for group 14/16 compounds,7 for instance.
The organic groups on the periphery of organogermanium and organotin sulfide complexes of the general type [(RfT)xSy] (Rf = functionalized organic ligand: R1 = CMe2CH2C(Me)O, HR2 = C2H4COOH; T = Ge, Sn; x/y = 4/6, 6/10) are reactive towards hydrazine derivatives, which allowed further derivatizations of the organic ligand shell.8 Hereby, an extension of the ligand shell towards ligands such as R3 = CMe2CH2C(Me)NNH2, R4 = {CMe2CH2C(Me)NNH}2CO, or R5 = CMe2CH2C(Me)NNC(2-py)2 was achieved. Reactions with transition metal compounds additionally enabled the extension of the inorganic core to form the organofunctionalized ternary clusters [(R3T)2(CuPR3)6S6],8 [(R2Sn)6(OMe)6Cu2S6]4−,8 [(R3Ge)4Pd6S12]9 and [R42{Sn(μ-S)2Cu(PPh2Me)}4].10 Bispyridine-decorated clusters [(R5Sn)4(ZnX)4S8] (X = Cl, Br, I) were obtained by slow diffusion of zinc halide solutions into solutions of the organotin sulfide cluster [(R5T)6S10].11
Further structurally characterized ternary group 11 metal/organotin sulfide clusters have so far been surrounded by unreactive organic groups, as in [(PhSn)2(CuPPh2Me)6S6],12 [(PhSn)12Cu19(PEt2Ph)3S28]+,13 and the mixed-valence compound [{(CH2)4SnIV}6(CuPPh3)6SnIICu4S12].14 Ternary complexes containing Ag/Sn/S or Au/Sn/S cores have not been characterized yet using X-ray diffraction.‡
Recently we reported new functional organotin sulfide complexes of the type [(R1,3Sn)3S4Cl] that exhibit defect heterocubane scaffolds with carbonyl and hydrazone groups.15 Herein, we show that these turned out to be suitable precursor complexes as well. [(R1Sn)3S4Cl] (A) reacts with the group 11 metal complexes [Cu(PPh3)3Cl], [Ag(PPh3)3Cl] and [Au(PPh3)Cl] in CH2Cl2 with (Me3Si)2S, in the case of Ag and Au complexes with subsequent in situ derivatization by hydrazine hydrate (Scheme 1), to yield single crystals of the following compounds comprising ternary clusters [(R1Sn)4(SnCl)2(CuPPh3)2S8]·4CH2Cl2 (1·4CH2Cl2), 1·[(R1Sn)4S6] (hereafter denoted as 2), [(R1Sn)4(SnCl)2(CuPPh3)2S8]·[(R1Sn)4S6] (3·[(R1Sn)4S6]), [(R3Sn)10Ag10S20]·3.5CH2Cl2 (4·3.5CH2Cl2), [{R1Sn(μ-S)}2{AuPPh3(μ-S)}2] (5), and [{R3Sn(μ-S)}2{AuPPh3(μ-S)}2]·5CH2Cl2 (6·5CH2Cl2). The clusters exhibit different topologies and compositions and comprise reactive substituents with either carbonyl or hydrazone groups. In addition, we report the single crystal X-ray diffraction analysis of (Me3Si)2S (see ESI†).
 | ||
| Scheme 1 Synthesis of compounds 1–6 (R1 = CMe2CH2C(Me)O, R3 = CMe2CH2C(Me)NNH2). | ||
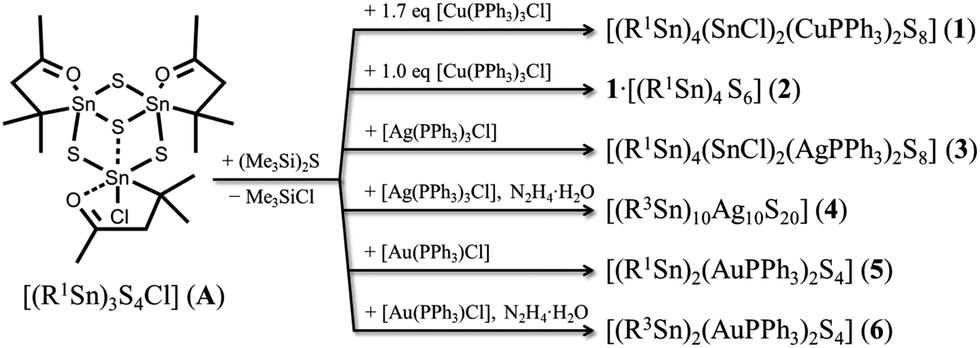
The reaction of A with 1.7 eq. [Cu(PPh3)3Cl] and (Me3Si)2S in CH2Cl2, and subsequent layering with n-hexane yielded [(R1Sn)4(SnCl)2(CuPPh3)2S8]·4CH2Cl2 (1·4CH2Cl2) as light yellow crystals. In the molecular structure of the cluster, two {R1Sn(μ-S)(μ3-S)}2 moieties are linked by two {(CuPPh3)(SnCl)} units (see Fig. 1). The R1Sn atoms Sn1 and Sn2 show a trigonal bipyramidal coordination environment, with the axial positions being occupied by O1, O2 and S1, whereas S2, S3 and C1 (Sn1) or S2, S4a and C7 (Sn2) are situated in the equatorial positions. The μ3-S-bridging atoms S1 and S4, and the μ-bridging atom S3 connect the {R1Sn(μ-S)(μ3-S)} moieties to the central part of the cluster. Here, Cu1 is surrounded in a tetrahedral fashion by a PPh3 ligand, S1, S4 and Sn3. The latter has a formal oxidation state of +II and exhibits a distorted pseudo-trigonal bipyramidal environment, with Cu1 and S3a in the “axial” positions (Cu1–Sn3–S3a 149.75(2)°), and Cl1, S4a and the sterically active lone pair in the “equatorial” positions; further pseudo-trigonal bipyramidal coordination geometries of SnII atoms have been described by Jurkschat and co-workers.16 The mixed-valence situation in 1 was confirmed using DFT calculations (see below). As another peculiarity, 1 comprises a rare Cu–Sn bond (Cu1–Sn3 2.6054(4) Å), which was formed in situ.17 It is not possible to monitor the formation process in detail due to the fairly complex reaction mixture and due to the poor solubility of the product. However, we assume that the reduction of RSnIV to SnII comes along with the oxidation of the released organic ligand as discussed for the mixed valence compounds [(R1,3Sn)3Se4][SnCl3] under similar reaction conditions;15 the oxidation of PPh3 or CuI, however, was not observed under the reaction conditions, which was clarified using NMR studies and test for Cu2+ with NH3.
 | ||
| Fig. 1 Molecular structure of 1 without H atoms (ellipsoids drawn at 50% probability); a = 1−x, −y, −z. Selected structural parameter [Å, °]: Sn–S 2.3654(8)–2.5058(8), Sn(1,2)–C 2.183(3)–2.192(3), Sn(1,2)⋯O(1,2) 2.516(2)–2.537(2), Sn3–Cl1 2.4713(8), Sn3–Cu1 2.6054(4), Cu1–S(1,4) 2.3626(9)–2.3166(8), Cu1–P1 2.2590(8); Sn–S–Sn 86.57(2)–89.70(3), S–Sn–S 91.24(3)–121.81(3), Cu1–Sn3–S3a 149.75(2), Cu1–Sn3–S4a 107.60(2), Cl1–Sn3–Cu1 111.23(2), Cl1–Sn3–S3a 90.20(3), Cl1–Sn3–S4a 95.70(3), C–Sn–O 72.10(10)–73.71(10). | ||
The observation of formal SnII and SnIV atoms is in agreement with natural charges, obtained by natural population analyses (NPA)18 of the DFT wave function, which was calculated by simultaneous optimization of the geometric and electronic structure using the program system TURBOMOLE.19 The formal SnIV atoms exhibit charges of +1.38 and +1.47, which are larger than the +0.88 charge calculated for the formal SnII atoms by a factor of 1.6 to 1.7. Details of the DFT calculations are provided in the ESI.†
Neither variation of the stoichiometry of the starting materials nor attempts to replace the Cl− ligand have led to the isolation of further clusters until now. An analogous reaction with 1.0 eq. [Cu(PPh3)3Cl], however, yielded 1·[(R1Sn)4S6] (2). The structural parameters of the two large co-crystallizing clusters in 2 are similar to those observed in 1·4 CH2Cl2 (see ESI†) and [(R1Sn)4S6].20
Upon reaction with [Ag(PPh3)3Cl] and (Me3Si)2S at −78 °C in CH2Cl2, and subsequent, careful warming-up to room temperature, a colorless precipitate was obtained, which was re-dissolved by addition of excess of CH2Cl2. Layering with n-hexane afforded [(R1Sn)4(SnCl)2(AgPPh3)2S8]·[(R1Sn)4S6] (3·[(R1Sn)4S6]) as light yellow crystals. The ternary cluster is isostructural to that in 2, hence the structural parameters are similar to those observed in 1 or 2, except the expected elongation of M–S, M–P and M–Sn bonds (Ag1–Sn3 2.6803(3) Å) for M = Ag in comparison with M = Cu, and a larger Sn–M–P angle (1: 115.40(3) for M = Cu, 3: 131.23(2) for M = Ag). For the selected structural parameters and NPA results see the ESI.†
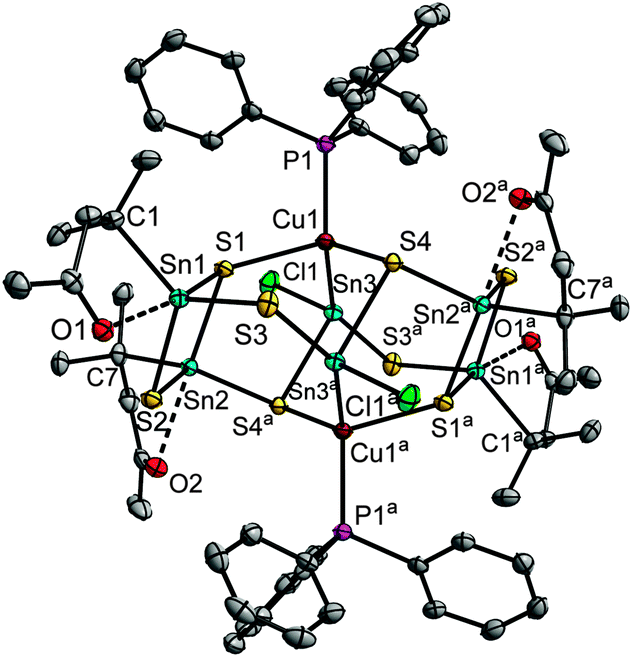
Addition of hydrazine hydrate to the re-dissolved solution, and subsequent layering with n-hexane produced orange crystals of the hydrazone functionalized cluster [(R3Sn)10Ag10S20]·3.5CH2Cl2 (4·3.5CH2Cl2). 4 consists of three {R3Sn(μ-S)(μ3-S)}2 units and two {[R3Sn(μ-S)]2(μ3-S)(μ4-S)} moieties that are linked by ten Ag atoms (see Fig. 2). All Sn atoms exhibit trigonal bipyramidal coordination, with N and S atoms in the axial positions. Four Ag atoms (Ag1, Ag5, Ag7, Ag10) are coordinated in a linear manner, while six other Ag atoms exhibit trigonal planar coordination. Closest Ag⋯Ag distances are in the range of 2.9353(7) Å to 3.3260(7) Å. Eight of the Ag atoms are connected this way, with a central butterfly-like arrangement and to adjacent Ag⋯Ag units, while one Ag⋯Ag unit is further apart. The cluster possesses pseudo-C2-symmetry. The pseudo-C2-axis runs through the centers of the Ag⋯Ag four-ring and the separate Ag⋯Ag dimer. In contrast to known silver sulfide clusters,1a the cluster does not additionally contain stabilizing phosphine groups attached to the silver atoms. N→Sn coordination of the bidentate organic ligands seems to provide sufficient kinetic stabilization.
 | ||
| Fig. 2 Molecular structure of the inorganic core of 4 along the pseudo-C2-axis (ellipsoids drawn at 50% probability, bonds to organic substituents are indicated by dashed lines). Ag⋯Ag contacts below 3.4 Å are drawn as dashed red lines. Selected structural parameters [Å, °]: Sn–S 2.3865(16)–2.5987(17), Sn–C 2.170(6)–2.187(7), Sn⋯N 2.362(5)–2.456(5), Ag–S 2.3666(16)–2.9040(16), Ag⋯Ag 2.9353(7)–3.3260(7); S–Sn–S 89.57(5)–122.29(6), S–Ag–S 83.47(5)–169.97(6), Sn–S–Sn 85.13(5)–89.69(5), Ag–S–Ag, 72.65(5)–146.30(6), Ag–S–Sn 80.15(5)–144.91(7), C–Sn–N 71.6(2)–75.8(2). | ||
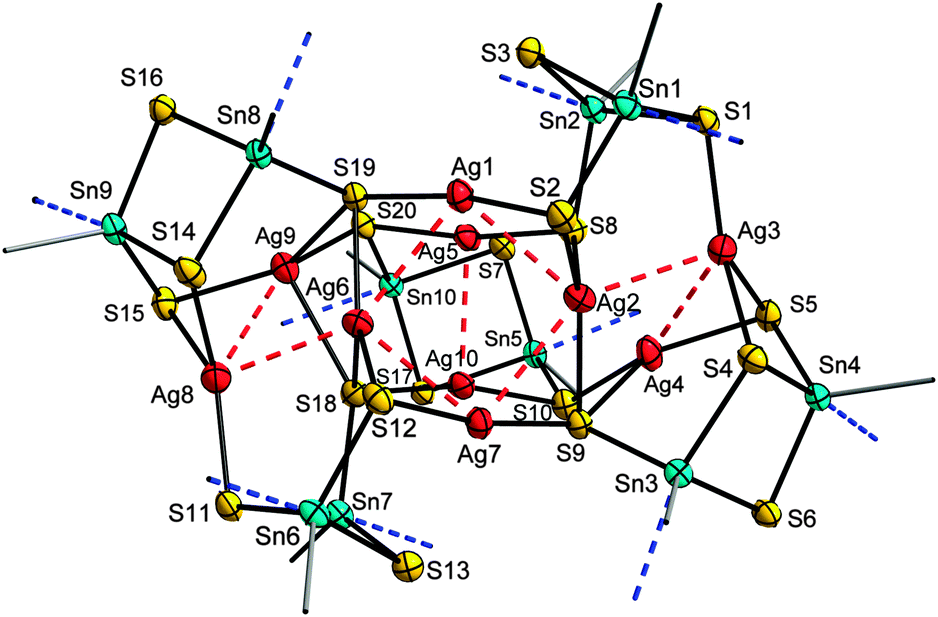
The employment of a related gold complex, [Au(PPh3)Cl], along with (Me3Si)2S in the reaction with A afforded single-crystals of the complex [{R1Sn(μ-S)}2{AuPPh3(μ-S)}2] (5). Moreover, addition of hydrazine hydrate to the reaction mixture yielded the hydrazone functionalized derivative [{R3Sn(μ-S)}2{AuPPh3(μ-S)}2]·5CH2Cl2 (6·5CH2Cl2) (see Fig. 3). In the present case, the derivatization of the organic ligand did not affect the molecular structure of the complex. In 5 and 6, a central {R1,3Sn(μ-S)}2 unit (cf.ref. 21) is terminated by two {Au(PPh3)(μ-S)} groups. As in 1–4, organo-decorated Sn atoms exhibit a trigonal bipyramidal coordination environment, while all Au atoms show linear coordination. The structural parameters of the central part is similar in both complexes, but the orientation of the attached {Au(PPh3)(μ-S)} units is different. In 5, they point away from the central Sn2S2 ring, with a C1–Sn1–S2–Au1 trans arrangement, whereas in 6, the corresponding atoms show a cis arrangement. While interatomic distances are similar in both complexes, the Sn1–S2–Au1 and S2–Au1–P1 angles show notable differences in both complexes, indicating the influence of the functional group included in the organic ligand.
 | ||
| Fig. 3 Molecular structures of 5 (left) and 6 (right) without H atoms (ellipsoids drawn at 50% probability). b = −x, 2−y, 1−z; c = 2−x, 1−y, −z. Selected structural parameters [Å, °]: 5: Sn–S 2.380(2)–2.463(2), Sn1–C1 2.189(8), Sn1⋯O1 2.714(6), Au1–S2 2.291(2), Au1–P1 2.260(2); Sn1–S1–Sn1b 86.30(7), S1–Sn1–S1b 93.70(7), Sn1–S2–Au1 96.63(9), S2–Au1–P1 178.29(9); 6: Sn–S 2.389(3)–2.525(3), Sn1–C1 2.186(14), Sn1⋯N1 2.479(11), Au1–S2 2.298(3), Au1–P1 2.250(3); Sn1–S1–Sn1c 89.06(11), S1–Sn1–S1c 90.94(11), Sn1–S2–Au1 103.86(13), S2–Au1–P1 172.84(12). | ||
In summary, a series of ternary group 11 organotin sulfide clusters have been synthesized and structurally characterized. The choice of the very group 11 metal M – although added as similar complexes [M(PPh3)xCl] (M/x = Cu/3, Ag/3, Au/1) – causes specific coordination environments that affect the molecular structures. Accordingly, the products exhibit different compositions and topologies. Unexpectedly, the reaction with the Cu and Ag precursor induced in situ reduction of one third of the Sn atoms under Sn–Cu and Sn–Ag bond formation. It was shown for the first time that a functionalized ternary M–Sn–S complex could undergo further derivatization.
This work was supported by the Deutsche Forschungsgemeinschaft within the framework of GRK1782.
Notes and references
- (a) O. Fuhr, S. Dehnen and D. Fenske, Chem. Soc. Rev., 2013, 42, 1871 RSC; (b) D. Fenske, T. Langetepe, M. M. Kappes, O. Hampe and P. Weis, Angew. Chem., Int. Ed., 2000, 39, 1857 CrossRef CAS.
- (a) C. B. Khadka, A. Eichhöfer, F. Weigend and J. F. Corrigan, Inorg. Chem., 2012, 51, 2747 CrossRef CAS PubMed; (b) C. Xu, J.-J. Zhang, Q. Chen, T. Duan, W.-H. Leung and Q.-F. Zhang, Inorg. Chem. Commun., 2012, 21, 1 CrossRef CAS PubMed; (c) A. Eichhöfer, J. Olkowska-Oetzel, D. Fenske, K. Fink, V. Mereacre, A. K. Powell and G. Buth, Inorg. Chem., 2009, 48, 8977 CrossRef PubMed.
- (a) D. Aldakov, A. Lefrançois and P. Reiss, J. Mater. Chem. C, 2013, 1, 3756 RSC; (b) S. Dehnen and M. Melullis, Coord. Chem. Rev., 2007, 251, 1259 CrossRef CAS PubMed.
- (a) C. B. Khadka, B. K. Najafabadi, M. Hesari, M. S. Workentin and J. F. Corrigan, Inorg. Chem., 2013, 52, 6798 CrossRef CAS PubMed; (b) W. Yu, O. Fuhr and D. Fenske, J. Cluster Sci., 2012, 23, 753 CrossRef CAS PubMed; (c) R. Langer, B. Breitung, L. Wünsche, D. Fenske and O. Fuhr, Z. Anorg. Allg. Chem., 2011, 637, 995 CrossRef CAS PubMed.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033 RSC.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- (a) M. Wagner, T. Zoller, W. Hiller, M. H. Prosenc and K. Jurkschat, Chem. Commun., 2013, 49, 8925 RSC; (b) M. Bouška, L. Střižík, L. Dostál, A. Růžička, A. Lyčka, L. Beneš, M. Vlček, J. Přikryl, P. Knotek, T. Wágner and R. Jambor, Chem. – Eur. J., 2013, 19, 1877 CrossRef PubMed; (c) B. E. K. Barth, B. A. Tkachenko, J. P. Eußner, P. R. Schreiner and S. Dehnen, Organometallics, 2014, 33, 1678 CrossRef CAS.
- Z. Hassanzadeh Fard, L. Xiong, C. Müller, M. Holynska and S. Dehnen, Chem. – Eur. J., 2009, 15, 6595 CrossRef PubMed.
- M. R. Halvagar, Z. Hassanzadeh Fard, L. Xiong and S. Dehnen, Inorg. Chem., 2009, 48, 7373 CrossRef CAS PubMed.
- M. R. Halvagar, Z. H. Fard and S. Dehnen, Chem. Commun., 2010, 46, 4716 RSC.
- B. E. K. Barth, E. Leusmann, K. Harms and S. Dehnen, Chem. Commun., 2013, 49, 6590 RSC.
- R. Hauser and K. Merzweiler, Z. Anorg. Allg. Chem., 2002, 628, 905 CrossRef CAS.
- A. Eichhöfer, J. Jiang, S. Lebedkin, D. Fenske, D. G. McDonald, J. F. Corrigan and C. Y. Su, Dalton Trans., 2012, 41, 3321 RSC.
- H. P. Nayek, W. Massa and S. Dehnen, Inorg. Chem., 2008, 47, 9146 CrossRef CAS PubMed.
- J. P. Eußner, B. E. Barth, E. Leusmann, Z. You, N. Rinn and S. Dehnen, Chem. – Eur. J., 2013, 19, 13792 CrossRef PubMed.
- (a) M. Wagner, C. Dietz, S. Krabbe, S. G. Koller, C. Strohmann and K. Jurkschat, Inorg. Chem., 2012, 51, 6851 CrossRef CAS PubMed; (b) Tin Chemistry: Fundamentals, Frontiers, and Applications, ed. A. G. Davies, M. Gielen, K. Pannell and E. Tiekink, Wiley, Hoboken, 2008 Search PubMed.
- Another example for the generation of formal SnII during cluster formation from a SnIV precursor was reported in ref. 14 for the compound [{(CH2)4SnIV}6(CuPPh3)6SnIICu4S12]. [(C4H7N2S)6SnCu4] also exhibits SnII–Cu bonds, see R. E. Allan, A. Bashall, J. S. Palmer, M. McPartlin, M. E. G. Mosquera, J. M. Rawson, A. E. H. Wheatleya and D. S. Wright, Chem. Commun., 1997, 1975 RSC.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Phys. Chem., 1985, 83, 735 CAS.
- (a) TURBOMOLE Version 6.2, © TURBOMOLE GmbH 2011. TURBOMOLE is a development of University of Karlsruhe and Forschungszentrum Karlsruhe 1989–2007; Ridft program: (b) K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652 CrossRef CAS; (c) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 CrossRef CAS ; BP86 functional: ; (d) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (e) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS PubMed; (f) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 33, 8822 CrossRef ; TZVP basis sets: ; (g) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (h) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC ; ECPs: ; (i) B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563 CrossRef CAS PubMed.
- Z. Hassanzadeh Fard, C. Müller, T. Harmening, R. Pöttgen and S. Dehnen, Angew. Chem., Int. Ed., 2009, 48, 4441 CrossRef PubMed.
- K. Jurkschat, S. van Dreumel, G. Dyson, D. Dakternieks, T. J. Bastow, M. E. Smith and M. Dräger, Polyhedron, 1992, 11, 2747 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further details of syntheses, analyses, and crystallographic data/further figures for 1–6 and (Me3Si)2S. CCDC 1011096–1011102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc05666c |
| ‡ X-ray crystallographic data: Data collection on a STOE IPDS2 diffractometer using graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) at 100 K. Structure solution and refinement by direct methods and full matrix least-squares on F2, respectively; SHELXTL software.22 |
| This journal is © The Royal Society of Chemistry 2014 |