
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning cation–anion interactions in ionic liquids by changing the conformational flexibility of the cation†
R. K.
Blundell
and
P.
Licence
*
School of Chemistry, The University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: peter.licence@nottingham.ac.uk
First published on 19th August 2014
Abstract
Cation–anion interactions can be tuned via the degree of cation constraint. X-ray Photoelectron Spectroscopy (XPS) experiments reveal that anion–cation based interactions may be enhanced by introducing conformational restriction into the substituent chains of quaternary ammonium cations. A larger degree of charge-transfer was observed for the constrained [C8C1Pyrr]+ cation relative to the [N6,6,6,14]+ cation.
Ionic liquids (ILs) are often referred to as “designer solvents”, due to the fact that by simply changing the component ions chemical and physical properties can be altered to suit a specific function.1 Therefore, one of the key goals in the field is to be able to predict how changes to the component ions will influence the properties of an ionic liquid system. To achieve this, ionic liquids must first be understood at a molecular level, and hence changes to the chemical structure and its effect upon inter-ion interactions must be explored. Thus far most ionic liquid research has focused upon cyclic nitrogen-based cations such as 1-alkyl-3-methylimidazolium, 1-alkyl-1-methylpyrrolidinium and 1-alkylpyridinium, and there are relatively few studies that address the distinction between cyclic cations and their acyclic counterparts, for example 1-alkyl-1-methylpyrrolidinium versus tetraalkylammonium.2–6 Herein, we describe a series of experiments designed to probe the influence of constrained and unconstrained cations upon cation–anion interactions utilising X-ray Photoelectron Spectroscopy (XPS).
XPS is now an established technique that can be used to monitor how changes to the chemical architecture of an IL may influence the electronic environment of the elements contained in the constituent ions, as well as providing quantitative information on the elemental composition of a sample.7 It has been suggested that “many (ionic liquid) applications could potentially profit from XPS analysis capabilities”.7 Most noticeably XPS of ionic liquids has entered a new domain with the advent of in situ reaction monitoring, the main advantage compared to other spectroscopic techniques being the simultaneous monitoring of all elements,8–10 thus exemplifying the power and value of this technique.
XPS investigations into cation–anion interactions have indicated a degree of charge-transfer from anion to cation which is correlated to anion basicity;11,12 however, little is known about the influence of the cation upon charge-transfer. In a recent XPS study upon tetraalkylammonium-based ILs and their phosphonium analogues we found that changing the cationic core from nitrogen to phosphorus had minimal impact upon charge-transfer due to a perceived hydrocarbon-based shielding of the cationic core.13 In this work we explore the concept of hydrocarbon-based shielding of the cationic core and its impact upon cation–anion interactions by comparison of a relatively unconstrained [N6,6,6,14]+ cation with a conformationally constrained [C8C1Pyrr]+ cation.
XP spectra were recorded using a Kratos Axis Ultra Spectrometer employing a monochromated Al Kα source (1486.6 eV), hybrid (magnetic/electrostatic) optics, hemispherical analyser and a multi-channel plate and delay line detector (DLD). Liquid samples were prepared by placing a small drop (≈10 mg) of ionic liquid onto a stainless steel multi-sample bar; solid samples were fixed to the bar using double-sided adhesive tape. All samples were pre-pumped in a preparative chamber to pressures lower than 10−6 mbar before transfer into the main analytical chamber to ensure removal of any volatile material. Charge referencing was achieved post-data collection by setting the Caliphatic 1s component to 285.0 eV, a full description of this procedure has been described elsewhere.14
The 1-octyl-1-methylpyrrolidinium and trihexyl(tetradecyl)ammonium cations are structurally very similar; however, a large proportion of the alkyl chain for [C8C1Pyrr]+ is effectively “pinned back” in a 5-membered heterocyclic ring. Subsequently, we expect the nitrogen core to be more exposed than in the case of [N6,6,6,14]+ in which the central nitrogen is shielded by a layer of conformationally flexible long-chain hydrocarbon. Crystallographic data reported by Adamova et al.15 supports this assertion since they describe the N⋯Cl distance for [N2,2,2,2]Cl as significantly longer than for [C3C1Pyrr]Cl; owing to the fact that the presence of strained rings around the central nitrogen atom for [C3C1Pyrr]Cl allows a closer approach of the halide anion.15
By consideration of the Coulomb potential, the interaction energy of two point charges is inversely proportional to separation distance. Consequently, the closer two point charges are to each other the stronger the interaction between them. Therefore, it is anticipated that a greater degree of charge transfer will occur in the case of the constrained [C8C1Pyrr]+ cation due to a stronger interaction with the anion, which would be reflected in the measured binding energy (BE) values for the anionic components.
The Cl 2p high resolution scan for each cation is presented in Fig. 1. It should be noted that due to spin–orbit coupling the photoelectron line is split into a doublet by a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (2p1/2:2p3/2). This is an inherent property of p-orbital photoelectron lines and does not indicate two different electronic environments of chlorine.
:2 (2p1/2:2p3/2). This is an inherent property of p-orbital photoelectron lines and does not indicate two different electronic environments of chlorine.
 | ||
| Fig. 1 High resolution XP spectra of the Cl 2p region for [N6,6,6,14]Cl and [C8C1Pyrr]Cl. Areas normalised to the area of Cl 2p photoemission peak for [N6,6,6,14]Cl. | ||
A 0.3 eV shift to higher binding energy is observed for the Cl 2p3/2 component of [C8C1Pyrr]Cl (Table 1, Fig. 1), this is greater than the experimental error (±0.1 eV) and indicates a noticeable change in the electronic environment of the anion, which we attribute to the availability of the cationic core. It should be noted that ΔBE(Cl 2p1/2–2p3/2) is 1.6 eV for each cation, this consistency reinforces that a true shift is observed and is not the result of other processes such as beam damage. A small broadening of the Cl 2p peaks is observed for the [N6,6,6,14]+ cation, which is likely to be a result of differences in surface charging of the samples. The increase in binding energy of the Cl 2p3/2 component for [C8C1Pyrr]Cl, relative to [N6,6,6,14]Cl, suggests that the chloride anion is more electron deficient when partnered with the 1-octyl-1-methylpyrrolidinium cation and indicates a greater degree of charge-transfer from anion to cation for this ionic liquid. This supports our proposal of a hydrocarbon-based shielding of the cationic core from the anion for cations containing long alkyl chains such as [N6,6,6,14]Cl.
| Compound | Binding energy/eV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cation | Anion | ||||||||||
| Ncation 1s | Caliphatic 1s | Cinter 1s | Chetero 1s | Cl 2p3/2 | B 1s | F 1s | Canion 1s | Nanion 1s | O 1s | S 2p3/2 | |
| [N6,6,6,14]Cl | 402.1 | 285.0 | 285.6 | 286.3 | 196.6 | — | — | — | — | — | — |
| [C8C1Pyrr]Cl | 402.1 | 285.0 | 285.5 | 286.1 | 196.9 | — | — | — | — | — | — |
| [N6,6,6,14][BF4] | 402.3 | 285.0 | 285.6 | 286.5 | — | 194.0 | 685.7 | — | — | — | — |
| [C8C1Pyrr][BF4] | 402.4 | 285.0 | 285.4 | 286.5 | — | 194.1 | 685.9 | — | — | — | — |
| [N6,6,6,14][NTf2] | 402.5 | 285.0 | 285.7 | 286.7 | — | — | 688.7 | 292.8 | 399.3 | 532.5 | 168.9 |
| [C8C1Pyrr][NTf2] | 402.7 | 285.0 | 285.6 | 286.8 | — | — | 688.8 | 292.9 | 399.5 | 532.7 | 169.0 |
A similar trend is also observed when comparing the F 1s high resolution scans for [C8C1Pyrr][BF4] and [N6,6,6,14][BF4] (Table 1, Fig. 2), in which a 0.2 eV shift is observed. The B 1s component also shifts slightly to higher binding energy by 0.1 eV for the [C8C1Pyrr]+ cation, however this is within the experimental error and so we can deduce that the cationic structure has minimal impact upon the electronic environment of the central boron atom. This may simply be the result of the inversely proportional relationship between interaction energy and separation distance, i.e. the fluorine atoms are closer to the cationic core and experience a stronger interaction, whereas the increased distance to the boron atom results in a weaker interaction.
 | ||
| Fig. 2 High resolution XP spectra of the F 1s region for [N6,6,6,14][BF4] and [C8C1Pyrr][BF4]. Areas normalised to the area of F 1s photoemission peak for [N6,6,6,14][BF4]. | ||
Furthermore, point charges calculated by de Andrade et al.16 and Tsuzuki et al.17 indicate that the negative charge of the [BF4]− anion is distributed across the four fluorine atoms, which could provide justification for the fact that changes in charge-transfer are more noticeable in the F 1s binding energies.
It has been previously shown that there is a strong correlation between the degree of charge-transfer and the solvatochromically determined hydrogen-bond basicity (β) of the anion; whereby high basicity anions such as halides transfer a significant amount of charge, with the opposite true for low basicity anions such as [NTf2]−.11,12 This could also provide an explanation for the smaller difference in F 1s binding energy (0.2 eV) than the observed Cl 2p3/2 shift of the chloride ionic liquids, as the [BF4]− anion naturally transfers less charge than the Cl− anion. To date, solvatochromic parameters have been extensively reported for 1-alkyl-3-methylimidazolium-based ionic liquids, whereas significantly fewer have been reported for 1-alkyl-1-methylpyrrolidinium and tetraalkylammonium-based ILs.18 Subsequently, a comparison of the Kamlet–Taft (β) parameter with Ncation 1s binding energy cannot be made in this study, although an analogous trend to the imidazolium based systems would be expected.
A comparison of binding energies for [C8C1Pyrr][NTf2] and [N6,6,6,14][NTf2] indicate that the anionic components all match within the experimental error excluding O 1s and N 1s (Table 1). The O 1s and N 1s binding energies are shifted to higher binding energy by 0.2 eV for [C8C1Pyrr][NTf2] in comparison to [N6,6,6,14][NTf2]. Examination of point charges determined by Hunt et al.,19 Canongia Lopes et al.20 and Tsuzuki et al.17 demonstrate that the negative charge is mainly distributed across the oxygen and nitrogen atoms of the [NTf2]− anion. Therefore, it is likely that the electronic environment and hence binding energies of these atoms will be most affected by changes to electron density, which can be seen in this study (Table 1).
The N 1s high resolution scan for [C8C1Pyrr][NTf2] and [N6,6,6,14][NTf2] is presented in Fig. 3. It can be seen that there are two distinguished nitrogen environments for each compound corresponding to cationic and anionic nitrogen. A shift to higher binding energy of anionic nitrogen for [C8C1Pyrr][NTf2] can clearly be seen.
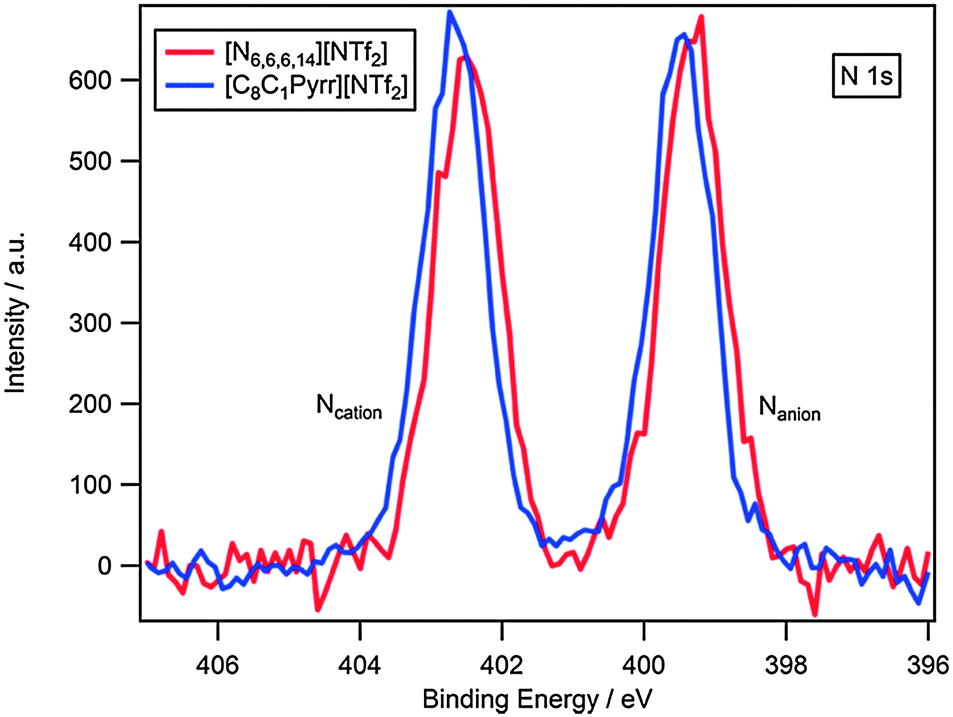 | ||
| Fig. 3 High resolution XP spectra of the N 1s region for [N6,6,6,14][NTf2] and [C8C1Pyrr][NTf2]. Areas normalised to the area of F 1s photoemission peak for [N6,6,6,14][NTf2]. | ||
Interestingly, the cationic nitrogen peak is also shifted 0.2 eV higher for [C8C1Pyrr][NTf2] relative to [N6,6,6,14][NTf2], which indicates that the electronic environment of Ncation for [N6,6,6,14][NTf2] is significantly more electron-rich than [C8C1Pyrr][NTf2].
To explain this observation the charge-transfer ability of the anion must be considered. It has been established that the Ncation 1s binding energy follows the trend [NTf2]− > [BF4]− > Cl−, which corresponds to an increase in charge transfer with increasing anion basicity;11 thus, the electronic environment of Ncation is more electron-poor when coupled with the [NTf2]− anion. Consequently, a stronger electron-releasing effect of the aliphatic chains will be induced for ionic liquids containing an [NTf2]− anion.
Considering that the [N6,6,6,14]+ cation has a substantially larger aliphatic contribution than [C8C1Pyrr]+, it seems reasonable that the Ncation environment for [N6,6,6,14][NTf2] experiences a stronger inductive effect from the surrounding alkyl chains causing a subsequent shift to lower binding energy.
In conclusion, the electronic environment of the anion is dependent upon cationic structure, with an increase in charge-transfer from anion to cation achieved for the more constrained structure of a 1-octyl-1-methylpyrrolidinium cation. This provides evidence for a hydrocarbon-based shielding of the cationic core for cations containing long-alkyl chains, which subsequently impacts upon cation–anion interactions. This research not only furthers our understanding of inter-ion interactions of ILs, but also provides a step towards being able to predict how changing the different components of an ionic liquid will impact upon their respective electronic environments, a step that is key to developing the ability to tune an ionic liquid system to meet a specific function.
We gratefully acknowledge the University of Nottingham and EPSRC (EP/K005138/1) for financial support and Ms Emily F. Smith for helpful discussions and critical advice.
Notes and references
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391 CrossRef CAS.
- H. Tokuda, K. Ishii, M. Susan, S. Tsuzuki, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 2833 CrossRef CAS PubMed.
- S. Tsuzuki, H. Matsumoto, W. Shinoda and M. Mikami, Phys. Chem. Chem. Phys., 2011, 13, 5987 RSC.
- T. M. Alam, D. R. Dreyer, C. W. Bielwaski and R. S. Ruoff, J. Phys. Chem. A, 2011, 115, 4307 CrossRef CAS PubMed.
- T. M. Alam, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, J. Phys. Chem. B, 2013, 117, 1967 CrossRef CAS PubMed.
- C. H. Xu, A. Durumeric, H. K. Kashyap, J. Kohanoff and C. J. Margulis, J. Am. Chem. Soc., 2013, 135, 17528 CrossRef CAS PubMed.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H.-P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158 CrossRef CAS PubMed.
- C. Kolbeck, I. Niedermaier, N. Taccardi, P. S. Schulz, F. Maier, P. Wasserscheid and H.-P. Steinrück, Angew. Chem., Int. Ed., 2012, 51, 2610 CrossRef CAS PubMed.
- I. Niedermaier, C. Kolbeck, N. Taccardi, P. S. Schulz, J. Li, T. Drewello, P. Wasserscheid, H.-P. Steinrück and F. Maier, ChemPhysChem, 2012, 13, 1725 CrossRef CAS PubMed.
- H. P. Steinruck, Phys. Chem. Chem. Phys., 2012, 14, 5010 RSC.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H.-P. Steinrück, Chem. – Eur. J., 2010, 16, 9018 CrossRef CAS PubMed.
- S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244 RSC.
- R. K. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278 RSC.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. L. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797 RSC.
- G. Adamova, R. L. Gardas, M. Nieuwenhuyzen, A. V. Puga, L. P. N. Rebelo, A. J. Robertson and K. R. Seddon, Dalton Trans., 2012, 41, 8316 RSC.
- J. de Andrade, E. S. Boes and H. Stassen, J. Phys. Chem. B, 2002, 106, 13344 CrossRef CAS.
- S. Tsuzuki, H. Tokuda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2005, 109, 16474 CrossRef CAS PubMed.
- P. G. Jessop, D. A. Jessop, D. B. Fu and L. Phan, Green Chem., 2012, 14, 1245 RSC.
- P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9 CrossRef CAS.
- J. N. Canongia Lopes, J. Deschamps and A. A. H. Padua, J. Phys. Chem. B, 2004, 108, 2038 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data and XP spectra. See DOI: 10.1039/c4cc05505e |
| This journal is © The Royal Society of Chemistry 2014 |