
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile access to a novel NHC-stabilized silyliumylidene ion and C–H activation of phenylacetylene†
Syed Usman
Ahmad
a,
Tibor
Szilvási
b and
Shigeyoshi
Inoue
*a
aInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: shigeyoshi.inoue@tu-berlin.de
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, 1111, Budapest, Hungary
First published on 22nd July 2014
Abstract
Taking advantage of two N-heterocyclic carbenes (NHCs), novel silyliumylidene ions 1a and 1b are prepared by a facile one-pot reaction of the corresponding dichlorosilanes with three equivalents of NHCs. For the first time, a C–H insertion reaction of phenylacetylene by a novel silyliumylidene ion is reported. The treatment of m-terphenyl substituted silyliumylidene ion 1a with three equivalents of phenylacetylene results in the formation of m-terphenyl substituted 1-alkenyl-1,1-dialkynylsilane 2.
Silylium ions [R3Si+], heavier analogues of carbenium ions, are among the strongest Lewis acids. It took a deliberate effort of over half a century towards their successful isolation.1 In general, the factors like specially designed non-coordinating counter anions, donor free solvents and kinetically stabilized bulky substituents were crucial towards the isolation of free silylium ions.2 The trivalent silicon centre in silylium ions is highly electrophilic and now Lewis acid catalysis as well as C–F bond activation are the most prominent applications of silylium ions.3 On the other hand, silylenes [R2Si:], heavier analogues of carbenes, have attracted much attention in the past 20 years and show interesting reactivities and potential applications in transition metal catalysis.4
Meanwhile, silyliumylidene ions [RSi:+] bear the best combined character of both silylium ions and silylenes. For example, the electrophilicity is more pronounced since the silicon centre possesses four valence electrons, two vacant orbitals and a lone pair of electrons. Consequently the isolation of silyliumylidene ions gets even more challenging.5 Several silyliumylidene cations, however, have been reported in the past ten years (Chart 1).6–11 This has been achieved either by employing well-designed ligands or multi-step synthetic methods. For example, the seminal work in this field is reported by Jutzi for silyliumylidene ion I, thanks to the stabilization effect of the pentamethylcyclopentadienyl ligand.6 Driess and co-workers utilized the intramolecular stabilization effect for the isolation of the silicon(II) cation II7 which also shows aromatic stabilization. The same group later on, reported on the synthesis of III8 and IV9 by the incorporation of especially designed bisiminophosphorane and bis N-heterocyclic carbene ligands, respectively. Moreover, So and coworkers reported on the synthesis of silyliumylidene ion V stabilized by DMAP and the amidinate ligand.10 It should also be mentioned that Filippou and co-workers reported on the striking example of silyliumylidene VI stabilized by two NHCs in a three step synthetic methodology starting from SiI4.11a
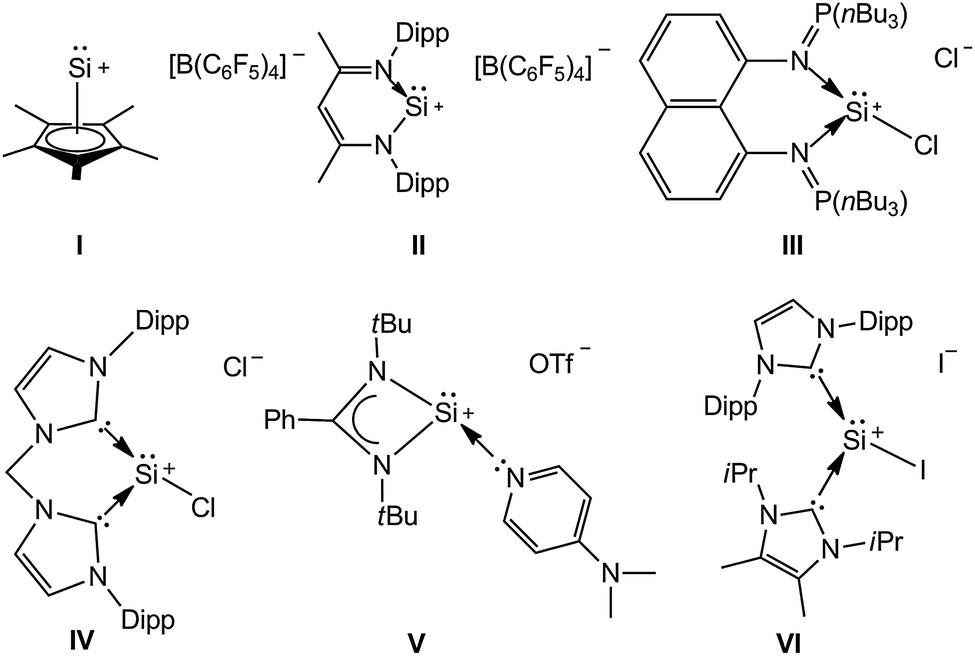 | ||
| Chart 1 Examples of isolable silyliumylidene ions. | ||
Despite their interesting character, the reactivity study of silyliumylidenes is still in its infancy.12 For example, the catalytic behavior of I in the controlled degradation of ether,13 the remarkable synthesis of a silylone from IV9 and activation of elemental sulfur from III8 and V10 remain as the highlights of the reactivity of silyliumylidene ions. Herein we report the C–H insertion in phenylacetylene using a novel silyliumylidene ion synthesized in a facile single step methodology.
The computational studies performed by Müller suggested a stable silyliumylidene ion substituted by the kinetically stabilizing terphenyl group at the silicon-center.14 This prompted us to employ the terphenyl substituent for the synthesis of a novel silyliumylidene ion. On the other hand, N-heterocyclic carbenes (NHCs) are employed as external donors to provide the desired stabilization for the low-valent silicon compounds.11,15–17 Thus, we employed two NHCs parallel to the methodology used by Filippou towards the isolation of NHC-stabilized chlorosilylene.16b Interestingly, we have found that the target m-terphenyl substituted silyliumylidene ion 1a stabilized by two NHCs can be obtained in a very facile one step experimental procedure (Scheme 1). In addition, we have also employed the triisopropylphenyl substituent at the silicon centre in order to generalize this convenient synthesis (Scheme 1). The synthetic methodology involves the addition of a solution of three equivalents of Me4NHC (Me4NHC = 1,3,4,5-tetramethylimidazol-2-ylidene) to a solution of the corresponding dichlorosilane m-TerSiHCl218 for the synthesis of 1a as well as TippSiHCl219 for the synthesis of 1b (m-Ter = 2,6-Mes2C6H3, Mes = 2,4,6-trimethylphenyl, Tipp = 2,4,6-triisopropylphenyl). Compound 1a is obtained by slow stirring of the reaction mixture only during the addition of the NHC at room temperature. Overnight standing of the reaction mixture results in the yellow-orange crystals of 1a which are obtained in 54% yield based on the 1H NMR spectrum. Compound 1b, however, is synthesized by slow addition of NHC to a heated solution of TippSiHCl2 in benzene at 50 °C. Consequently, the solution is separated from the imidazolium salt and the solvent is reduced in volume and allowed to stand overnight at room temperature. Compound 1b is obtained as a bright yellow crystalline product in 45% yield.
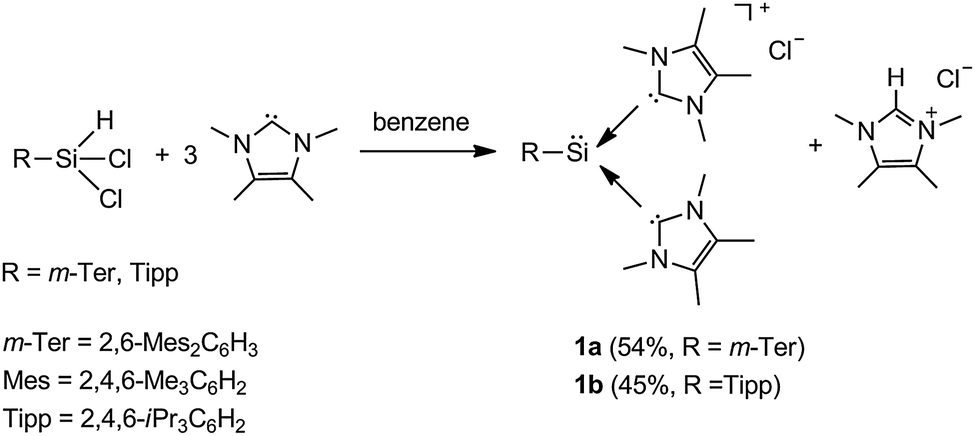 | ||
| Scheme 1 Synthesis of silyliumylidenes 1a and 1b. | ||
The 1H NMR spectrum of 1a and 1b at room temperature displays one set of signals for the respective m-terphenyl and Tipp groups as well as the two coordinated NHCs. Additionally, for 1a, four broad singlets (corresponding to the N-Me and C-Me protons of NHCs, the 3,5-Mes protons and the ortho-Me protons of the mesityl group) observed at room temperature split into two singlets each at −20 °C. On the other hand, the 1H NMR spectrum of 1b does not show a similar broadening of the chemical shifts as seen for 1a. The 13C NMR resonances for the carbene-carbons of 1a and 1b are observed at 160.3 ppm and 159.7 ppm as singlets, in the 13C NMR spectrum, respectively. One sharp signal was observed at −68.85 ppm for 1a in the 29Si NMR spectrum, whereas the 29Si NMR spectrum of 1b displayed a chemical shift at −69.50 ppm as a singlet. These are downfield shifted compared to V (δ = −82.3 ppm),10 but upfield shifted compared to IV (δ = −58.4 ppm).9 The calculated values of 29Si NMR resonances of 1a and 1b (δ = −67.32 ppm and −68.55 ppm, respectively, B3LYP/6-31G(d)[C,N,H]/6-311G(3d)[Si]) are in good agreement with the experimental values.
Compound 1a crystallizes in the monoclinic space group P21/c as separated ion pairs (the shortest Si–Cl distance is 6.234 Å). The molecular structure of 1a is depicted in Fig. 1. The silicon centre is three fold coordinated to the two N-heterocyclic carbenes and ipso carbon (C1) of the m-terphenyl group. The sum of the bond angles around the Si1 atom is 310.2°. The Si–C bond distances in 1a for the coordinated NHCs are almost identical [1.948(19) and 1.967(19) Å] to the Si–C bonds of the coordinated NHCs in VI [SiI(NHCiPr2Me2)(NHCdipp)I]I [1.947(2) and 1.967(2) Å] for Si–C (NHCdipp) and Si–C (NHCiPr2Me2), respectively (NHCiPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene, NHCdipp = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene).11a
 | ||
| Fig. 1 Molecular structure of 1a. Thermal ellipsoids represent the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Si(1)–C(1) 1.9355(19), Si(1)–C(32) 1.9481(19), Si(1)–C(25) 1.9665(19), C(1)–Si(1)–C(32) 105.06(8), C(1)–Si(1)–C(25) 111.36(8), and C(32)–Si(1)–C(25) 93.78(8). | ||
Scheme 2 presents the possible mesomeric structures of compounds 1a and 1b. A donor–acceptor stabilized silicon(II) cation is the mesomeric structure based on the high degree of pyramidalization observed for 1a (310.21).20 The other mesomeric form is the zwitterionic structure, where the positive charge is dispersed over the two NHC backbones.
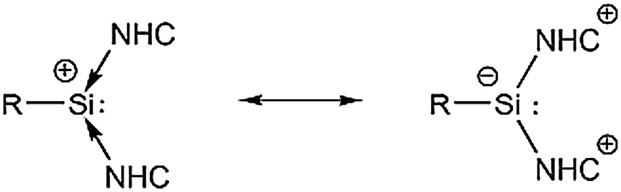 | ||
| Scheme 2 Zwitterionic and donor–acceptor stabilized canonical structures of 1a (R = m-Ter) and 1b (R = Tipp). | ||
Furthermore, DFT calculations for the cationic part of 1a and 1b were carried out at B3LYP/6-31G(d) level of theory. The HOMO of 1a shows mainly the lone pair orbital at the silicon centre, whereas the LUMO of 1a is dispersed over the NHC skeletons (Fig. 2). Molecular orbitals of 1b are similar to that of 1a (ESI†). The NBO charge clearly shows that the silicon centre bears positive net charge (+0.798 for 1a and + 0.804 for 1b).
 | ||
| Fig. 2 Molecular orbitals of 1a, the HOMO (left, −6.92 eV) and the LUMO (right, −3.37 eV). | ||
Silylenes undergo cycloaddition reaction with internal alkynes21,22 and terminal alkynes22 as well as activate C–H bonds in terminal alkynes17,23. It is of note that Müller has predicted the potential of silyliumylidene ions towards C–H activation through the generation of a silylium ion by proton abstraction from solvent by an intermediate silyliumylidene.24 We have therefore embarked on reactivity investigation of the silyliumylidene 1a with the terminal alkyne. Interestingly, the unprecedented reactivity of the silyliumylidene 1a towards phenylacetylene was observed. The reaction of 1a with three equivalents of phenylacetylene in acetonitrile yields the m-terphenyl substituted 1-alkenyl-1,1-dialkynylsilane 2 in 68% yield, solely as the Z-isomer (Scheme 3). Compound 2 was fully characterized by multinuclear NMR spectrometry, ESI-HRMS as well as single crystal X-ray analysis. The 1H-NMR of 2 displays one set of signals for the m-terphenyl group, the alkynyl substituents and the alkenyl substituent. The ethylene (Si–CH = CH–Ph) protons are observed as doublets at 5.16 and 6.97 ppm. In the 13C NMR spectrum of 2, the ethylyne carbon resonances (Si–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph) appear at 90.9 and 106.7 ppm, whereas the ethylene carbon chemical shifts (Si–CH = CH–Ph) are observed at 126.5 and 145.3 ppm. The 29Si-NMR chemical shift of 2 is observed at −62.28 ppm as a sharp singlet, which is downfield shifted in comparison to that of precursor 1a (δ = −68.85 ppm). In addition, this value fits well with the calculated value (δ = −59.02 ppm, B3LYP/6-31G(d)[C,N,H]/6-311G(3d)[Si]). Furthermore, DFT calculations [RI-B97-D/cc-pVTZ(SMD = acetonitrile)//RI-B97-D/6-31G* level of theory] were performed to suggest a plausible mechanism for the formation of 2 as a Z-isomer as the sole product (see ESI†). According to the DFT calculations, the formation of the E-isomer requires high activation barriers and therefore the formation of the E-isomer is kinetically not favored (for details see also the ESI†).
C–Ph) appear at 90.9 and 106.7 ppm, whereas the ethylene carbon chemical shifts (Si–CH = CH–Ph) are observed at 126.5 and 145.3 ppm. The 29Si-NMR chemical shift of 2 is observed at −62.28 ppm as a sharp singlet, which is downfield shifted in comparison to that of precursor 1a (δ = −68.85 ppm). In addition, this value fits well with the calculated value (δ = −59.02 ppm, B3LYP/6-31G(d)[C,N,H]/6-311G(3d)[Si]). Furthermore, DFT calculations [RI-B97-D/cc-pVTZ(SMD = acetonitrile)//RI-B97-D/6-31G* level of theory] were performed to suggest a plausible mechanism for the formation of 2 as a Z-isomer as the sole product (see ESI†). According to the DFT calculations, the formation of the E-isomer requires high activation barriers and therefore the formation of the E-isomer is kinetically not favored (for details see also the ESI†).
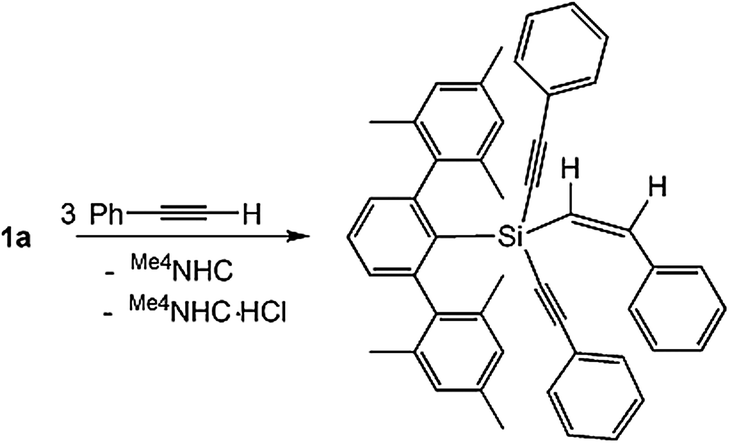 | ||
| Scheme 3 Reaction of 1a with three equivalents of phenylacetylene. | ||
Compound 2 crystallizes in the monoclinic space group C2/c and the molecular structure is shown in Fig. 3. The central silicon possesses a distorted tetrahedral geometry with two alkylyne groups (C33–C34 and C41–C42) and one alkylene group (C25–C26) terminally coordinated to the silicon, whereas the maximum steric room is occupied by the umbrella shaped m-terphenyl substituent on the silicon. The broadest angle at the Si centre is displayed between C(terphenyl)–Si–C(alkylene) as 115.03°. The alkylene substituent (C25–C26) reveals Z-configuration at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in contrast to the E-configuration of 1-alkenyl-1-alkynylsilole reported by Cui and co-workers.17 Another feature to be compared with the 1-alkenyl-1-alkynylsilole is the Si–C bond lengths. While the Si–C(alkynyl) bonds (1.82(15) Å and 1.83(15) Å) of 2 are similar to those of the 1-alkenyl-1-alkynylsilole (1.83(2) Å), however the Si–C(alkenyl) bond (1.87(25) Å) is slightly longer than that of 1-alkenyl-1-alkynylsilole (1.83(2) Å) which leads back to electronic interaction of silicon and butadiene in the silole ring.17
C double bond in contrast to the E-configuration of 1-alkenyl-1-alkynylsilole reported by Cui and co-workers.17 Another feature to be compared with the 1-alkenyl-1-alkynylsilole is the Si–C bond lengths. While the Si–C(alkynyl) bonds (1.82(15) Å and 1.83(15) Å) of 2 are similar to those of the 1-alkenyl-1-alkynylsilole (1.83(2) Å), however the Si–C(alkenyl) bond (1.87(25) Å) is slightly longer than that of 1-alkenyl-1-alkynylsilole (1.83(2) Å) which leads back to electronic interaction of silicon and butadiene in the silole ring.17
 | ||
| Fig. 3 Molecular structure of 2. Thermal ellipsoids represent 50% probability level. Hydrogen atoms (except those on C25 and C26) are omitted for clarity. Selected bond lengths [Å] and angles [°]: Si(1)–C(33) 1.8281(15), Si(1)–C(41) 1.8320(16), Si(1)–C(25) 1.8713(15), Si(1)–C(1) 1.8913(15), C(33)–Si(1)–C(41) 106.92(7), C(33)–Si(1)–C(25) 107.83(7), C(41)–Si(1)–C(25) 103.26(7), C(33)–Si(1)–C(1) 112.34(6), C(41)–Si(1)–C(1) 110.80(7), and C(25)–Si(1)–C(1) 115.03(7). | ||
In conclusion, we report on the synthesis of novel m-terphenyl and Tipp substituted silyliumylidene ions 1a and 1b stabilized by two NHCs through a facile synthetic route. This striking one pot reaction will be further generalized by employing various substituents at the silicon center in our laboratories. In addition, the promising reactivity of 1a is evident from its reaction with phenylacetylene leading to the C–H insertion product 2. Both compounds 1a and 2 are characterized by multinuclear NMR as well as single-crystal X-ray diffraction analysis. Moreover, DFT calculations are performed to establish the energy pathway for the formation of 2 from 1a, which rationalizes the formation of the Z-isomer in this reaction. Further reactivity studies on 1a and 1b are ongoing and will be reported in due course.
We are exceptionally grateful to the Alexander von Humboldt foundation (Sofja Kovalevskaja Program) for financial support. We thank Dr Elisabeth Irran for structural refinement of 1 and 2. We wish to thank Dr S. Kemper for helpful discussion. T. S. is thankful for the support of The New Széchenyi Plan TAMOP-4.2.2/B-10/1-2010-0009.
Notes and references
-
(a) J. Y. Corey, J. Am. Chem. Soc., 1975, 97, 3237 CrossRef CAS
; (b) J. B. Lambert, Y. Zhao and S. Zhang, J. Phys. Org. Chem., 2001, 14, 370 CrossRef CAS
-
(a) K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Müller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825 CrossRef CAS PubMed
-
(a) H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176 RSC
-
(a) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef CAS PubMed
- For transition metal stabilized silyliumylidenes, see
(a) N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958 CrossRef CAS PubMed
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849 CrossRef CAS PubMed
- M. Driess, S. Yao, M. Brym and C. van Wüllen, Angew. Chem., Int. Ed., 2006, 45, 6730 CrossRef CAS PubMed
- Y. Xiong, S. Yao, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 10074 CrossRef CAS PubMed
- Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147 CrossRef CAS PubMed
- H.-X. Yeong, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Chem. – Eur. J., 2013, 19, 11786 CrossRef CAS PubMed
-
(a) A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974 CrossRef CAS PubMed
-
(a) P. Jutzi, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller, H.-G. Stammler and A. B. Rozhenko, J. Am. Chem. Soc., 2009, 131, 12137 CrossRef CAS PubMed
- K. Leszczynska, A. Mix, R. J. F. Berger, B. Rummel, B. Neumann, H.-G. Stammler and P. Jutzi, Angew. Chem., Int. Ed., 2011, 50, 6843 CrossRef CAS PubMed
- T. Müller, Organometallics, 2010, 29, 1277 CrossRef
-
(a) S. Inoue and C. Eisenhut, J. Am. Chem. Soc., 2013, 135, 18315 CrossRef CAS PubMed
-
(a) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687 CrossRef CAS PubMed
- Y. Gao, J. Zhang, H. Hu and C. Cui, Organometallics, 2010, 29, 3063 CrossRef CAS
- R. S. Simons, S. T. Haubrich, B. V. Mork, M. Niemeyer and P. P. Power, Main Group Chem., 1998, 2, 275 CrossRef CAS
- See the ESI†.
- See detailed analysis in the ESI†.
-
(a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed
- S. Yao, C. van Wüllen, X. Sun and M. Driess, Angew. Chem., Int. Ed., 2008, 47, 3250 CrossRef CAS PubMed
- H. Cui, B. Ma and C. Cui, Organometallics, 2012, 31, 7339 CrossRef CAS
- C. Gerdes, W. Saak, D. Haase and T. Müller, J. Am. Chem. Soc., 2013, 135, 10353 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data and DFT calculations. CCDC 993048, 1a; 1000427, (2). For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc05181e |
| This journal is © The Royal Society of Chemistry 2014 |