
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis, coating and morphogenesis of conductive polymer materials through spontaneous generation of activated monomers†
Ryo
Muramatsu
a,
Yuya
Oaki
*a,
Kento
Kuwabara
a,
Kosei
Hayashi
b and
Hiroaki
Imai
*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: oakiyuya@applc.keio.ac.jp; hiroaki@applc.keio.ac.jp
bTokyo Metropolitan Industrial Technology Research Institute, 2-4-10 Aomi, Koto-ku, Tokyo 135-0064, Japan
First published on 13th August 2014
Abstract
Synthesis, coating, and morphogenesis of conductive polymers were achieved on a variety of substrates through spontaneous generation of activated monomer vapors under ambient pressure and low temperature conditions. The present approach facilitates the generation of complex hierarchical morphologies and the conductive coating for improvement of electrochemical properties.
Morphologies of materials are significant for their wide range of applications.1 Exploration of new methodologies is required to achieve fine tuning of morphologies in organic and inorganic materials. Morphologies of crystals from nanoscopic to macroscopic scales can be controlled by tuning the growth conditions.2 Design, synthesis, and self-assembly of functional organic molecules have been widely studied to generate the organized structures with controlled arrangement and orientation.3 However, the hierarchical morphology control of polymer materials, particularly in conductive polymers, has not been fully explored in previous studies.4–7 Here we focus on morphologies of conductive polymer materials from nanometer to millimeter scales. In general, it is not so easy to control polymerization reactions and morphogenesis simultaneously in the solution processes. In the present study, we have developed an approach for solvent-free synthesis, coating, and morphogenesis of conductive polymers through spontaneous generation of the activated monomers under ambient pressure and low temperature.
Coating and casting of materials with the assistance of templates are used for the morphogenesis in a wide range of length scales.4 Morphologies of conductive polymer materials have been controlled by using templates.5–8 Self-assembled molecules are used as molecular templates.6 Host materials with nanospace, such as porous materials, are used for the morphology control at the nanoscale.7 We developed a method for the hierarchical morphology replication from the original materials to conductive polymers by means of mesocrystals.8 However, the morphological variety was limited to the combinations of templates and polymers. If coating and casting of conductive polymers are achieved on a variety of substrates including templates, the morphological variety can be expanded. Therefore, we focused on a solvent-free approach for coating and casting of conductive polymers by using the monomer vapors and the oxidant crystals under ambient pressure and low temperature.
Vapor-phase polymerization was performed in a couple of systems. Polyethylene and polypropylene were produced on an industrial scale from the monomer vapors in contact with a powder of the catalyst.9 If conductive polymers are synthesized from the vapors of the monomers, the coating and casting on a variety of substrates including templates can be achieved by the diffusion of the monomer vapors. Previous studies showed the synthesis of conductive polymers from the monomer vapors.10–13 The vacuum deposition of the monomers and their polymerization were performed on a clean crystal surface.10 The oxidative chemical vapor deposition provided the conductive polymers from vapors of both the monomers and the oxidative agents on a variety of substrates.11 However, these methods were performed under vacuum conditions using specific equipment. In the vapor phase polymerization approaches,12 the diffusion of monomer vapors into a polymer matrix containing an oxidative agent led to the formation of the composites consisting of the matrix and the conductive polymer. The initial coating of oxidants is required for the polymer deposition prior to the exposure to the monomer vapors. In our present approach, both the initial coating of the oxidants and the vacuum conditions are not needed for synthesis, coating, and morphogenesis of conductive polymer materials. The activated monomer vapors were spontaneously generated from the original monomer vapors in contact with the oxidant crystals. Therefore, the polymerization and coating proceeded spontaneously on a variety of unmodified bare substrates under ambient pressure and low temperature conditions. In a previous work, the designed and synthesized unstable monomer vapors facilitated the deposition of a conductive polymer.13 In the present study, the spontaneous generation of the activated monomers facilitated the synthesis, coating, and morphogenesis of conductive polymers under ambient pressure and low temperature conditions.
Fig. 1 shows the experimental setup and the results. The powder of the oxidant crystals and the neat liquid of the monomers were separately placed in a sample bottle (Fig. 1a). For example, pyrrole (Py, C4H5N) and copper(II) nitrate trihydrate (Cu(NO3)2·3H2O) were adopted as the monomer and the oxidant crystals, respectively. The reaction was started at 60 °C after sealing the sample bottle. The color of the oxidant crystals changed to black. The color change suggested the formation of the polymerized materials on the crystals. Moreover, the polymerized materials black in color were observed on the walls of the sample bottle (Fig. 1b–d). Therefore, the substrates, such as a glass slide and a single crystal of alkali halides, were put in the sample bottle to study the deposition behavior (Fig. 1a). The polymerized materials black in color were deposited on these substrates. A flat film 200 nm in thickness was observed throughout the single crystal of potassium bromide (KBr) after deposition for 12 h (Fig. 1e and f). After deposition for 60 h, a textured morphology consisting of a wrinkled film 800 nm in thickness was obtained throughout the substrate (Fig. 1g). The resultant films were assigned to PPy by Fourier-transform infrared (FT-IR) spectroscopy (Fig. 1h). The results imply that conductive polymers, such as PPy, can be synthesized and coated on substrates by the present solvent-free approach from the monomer vapors in the presence of oxidant crystals.
 | ||
Fig. 1 Synthesis and coating of PPy on the substrates in the presence of Cu(NO3)2·3H2O. (a) The schematic illustration of the experimental setup. (b–d) The time-course observations of the sample bottle at 0 h (b), 2 h (c), and 24 h (d). (e) KBr crystal as a substrate before polymerization (upper panel) and its optical microscopy (OM) images (bottom panel). (f, g) The surface OM images (upper panels) and the cross-sectional FESEM images (bottom panels) of PPy deposited on a KBr single crystal substrate after the reaction for 12 h (f) and 60 h (g). (h) FT-IR spectrum of the PPy formed on a KBr crystal. The peaks of FT-IR spectrum are assigned to the following vibrations of PPy: C–H in-ring out-of-plane bending (A), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (B), C–C in-ring stretching (C), C–H stretching (D), and N–H stretching (E). C stretching (B), C–C in-ring stretching (C), C–H stretching (D), and N–H stretching (E). | ||
The oxidant crystals, substrates, and monomers were changed to study the polymerization and coating behavior. The thickness of the films was increased with an increase in the deposition time (Fig. 2). The deposition rate of PPy was increased upon changing the oxidant crystal from Cu(NO3)2·3H2O to iron(III) nitrate nonahydrate (Fe(NO3)2·9H2O) (Fig. 2). When other copper salts were used as the reference, polymerization and coating were not observed on the substrates (Fig. S1 in the ESI†). The deposition rate and the morphology variation of PPy were not influenced by changing the substrate from a KBr single crystal to a glass slide (Fig. 2 and Fig. S1 in the ESI†). The PPy flat films were formed on different substrates, such as single crystals of silicon (Si) and potassium chloride (KCl). In contrast, the wrinkled films showed a variety of patterned morphologies depending on the substrates (Fig. S2 in the ESI†). The formation process of the PPy films was observed by field emission scanning electron microscopy (FESEM) and field emission transmission electron microscopy (FETEM) (Fig. S3 in the ESI†). At the initial stage of the deposition, the nanoparticles were observed on the flat film. The size of the nanoparticles increased with an increase in the deposition time. The film thickness increased with the growth of the nanoparticles. A further increase in the reaction time induced the formation of the wrinkled films (Fig. 1g). The wrinkles of the films were induced by the internal stress with an increase in the particle size (Fig. S2 in the ESI†). The poly(3,4-ethylenedioxythiophene) (PEDOT) films were obtained by changing the monomer from Py to 3,4-ethylenedioxythiophene (EDOT) (Fig. S4 in the ESI†). The thickness of the PEDOT films was controlled by the reaction time. In contrast, the polymer films of poly(3-hexylthiophene) (P3HT) and polythiophene (PTp) were not formed on the substrates by the present approach.
 | ||
| Fig. 2 Relationship between the deposition time and the film thickness for coating of PPy in the presence of Cu(NO3)2·3H2O (a) and Fe(NO3)2·9H2O (b) on glass (the circles and diamonds) and KBr (the triangles and squares) substrates. The opened and filled symbols represent the flat and wrinkled films, respectively. | ||
The conductive polymers, such as PPy and PEDOT, were deposited not only on the crystal surface of the oxidants but also on the separately-placed substrates, although the neat liquid of the monomers and the crystals of the oxidative agents were separately placed in the sample bottle. In the vapor phase, the presence of pyrrole (Py) monomer and nitropyrrole (Py-NO2) was detected by gas chromatography mass spectroscopy (GC-MS) (Fig. S5 in the ESI†). The spontaneous generation of the activated monomers, namely nitropyrrole (Py-NO2) plays an important role in the polymerization in the present approach (Fig. S6 in the ESI†). The diffusion and adsorption of these monomer vapors initiate the polymerization on the substrates. The polymerization and coating mechanisms were studied by the formation of PPy in the presence of Cu(NO3)2·3H2O as the model case. The oxidative polymerization of Py proceeds on the crystal surface with the reduction of the copper ions in the oxidant crystal (Fig. S7 in the ESI†). Simultaneously, the nitrate is eliminated from the copper-related compounds. In the resultant PPy films, the presence of copper-related compounds was not detected by energy dispersive X-ray (EDX) analysis (Fig. S8 in the ESI†). These results imply that the spontaneous generation of Py-NO2 induced the synthesis and coating of PPy on the substrates and walls (Fig. S5 and S6 in the ESI†). Therefore, the volatile and activatable heteroaromatic monomers are required for the present approach. The oxidant crystals consisting of a reducible metal cation and an anion as an electron withdrawing group are suitable for the promotion of the reaction and polymerization.
If the polymers are coated in the pore spaces of template materials by the present approach, the casting and the subsequent morphology replication can be achieved. The substrate was changed to a one with a complex hierarchical morphology from nanoscopic to macroscopic scales. A sea urchin spine was adopted as a model of the template. The hierarchical morphology of a sea urchin spine was replicated to PPy (Fig. 3). A sea urchin spine has hierarchically organized structures consisting of magnesium-doped calcium carbonate (calcite).14 The spicular shape consisted of the sponge structure at the micrometer scale (Fig. 3a and b). The sponge skeletal body has a mesocrystal structure consisting of the oriented nanocrystals 20–50 nm in size (Fig. 3b and c).14 A sea urchin spine was set as a substrate in the sample bottle. The composite of the calcite and PPy was obtained after reaction for 60 h (Fig. S9 in the ESI†). The polymerization and coating proceeded not in the pores of the micrometer-scale sponge but in the skeletal body consisting of the nanocrystals. A hierarchical morphology of PPy was obtained after the dissolution of the original calcite by hydrochloric acid (Fig. 3d–g). The replicated PPy showed the micrometer-scale sponge structure (Fig. 3d and e). The flat surface without a grain boundary can be observed in the magnified image (Fig. 3f). The assembly of the PPy sheets can be observed in the FETEM image (Fig. 3g). The monomers were supplied from the vapor to the nanospace of mesocrystals. The polymerization and coating started from the crystal surface in the nanospace.8 As the reaction time increased, the nanospace was filled with PPy. The morphology replication was achieved by casting. If the substrates have the hierarchical structures with both the macroscopic morphologies and the nanospace, the morphogenesis of conductive polymers can be achieved by the present approach.
 | ||
| Fig. 3 Morphology replication through the casting by PPy. (a–c) FESEM images of the original sea urchin spine. (d–g) FESEM (d–f) and FETEM (g) images of the replicated architecture after the dissolution of the original calcite. | ||
The present technique was applied to conductive coating of an electrode active material for lithium-ion batteries (Fig. 4). In general, a conductive coating on an active material contributes to the improvement of the electrochemical performances as follows: formation of conductive passes for electrons with permeability of lithium ions, shortening of the electron transportation length with a decrease in the grain boundary, the improvement of the chemical stability during the reactions, and inhibition of the grain growth in the thermal treatment.15c Lithium titanate with the spinel structure (Li4Ti5O12), as an anode active material, was used to study the effect of the conductive coating. The powder of commercial Li4Ti5O12 nanocrystals 20–100 nm in size was set as a substrate with a neat liquid of Py and Fe(NO3)2·9H2O. The PPy coating was performed on the Li4Ti5O12 nanocrystals for 3 h. The thin amorphous layer 1 nm in thickness was partially formed on the nanocrystals (Fig. S10 in the ESI†). The content of the PPy was estimated to be 1.7 wt% in the composite (Fig. S10 in the ESI†). PPy was partially coated on the surface of the Li4Ti5O12 nanocrystals. The charge–discharge reactions were performed on the PPy-coated Li4Ti5O12 nanocrystals in the potential range of 1.0–2.7 V (versus Li/Li+). The typical charge–discharge curves of Li4Ti5O12 with the plateaus at 1.5 V vs. Li/Li+ were observed (Fig. 4a).15 After the PPy coating, the overpotential on the charge and discharge reactions, corresponding to the deviations of the potentials from 1.5 V vs. Li/Li+, was decreased. The current density of the charge and discharge reactions was increased from 0.2 C to 50 C (1 C = 0.175 A g−1). The PPy-coated Li4Ti5O12 nanocrystals showed improved specific capacity at a higher current density (Fig. 4b). A partial coating on the surface of the Li4Ti5O12 nanocrystals facilitates the improved electron conductivity and the smooth reaction with lithium ions. The electrochemical performances are ascribed to the improvement of the electron conductivity by the PPy coating at the nanoscale. The present technique can be used for a variety of conductive coatings on electrode active materials.
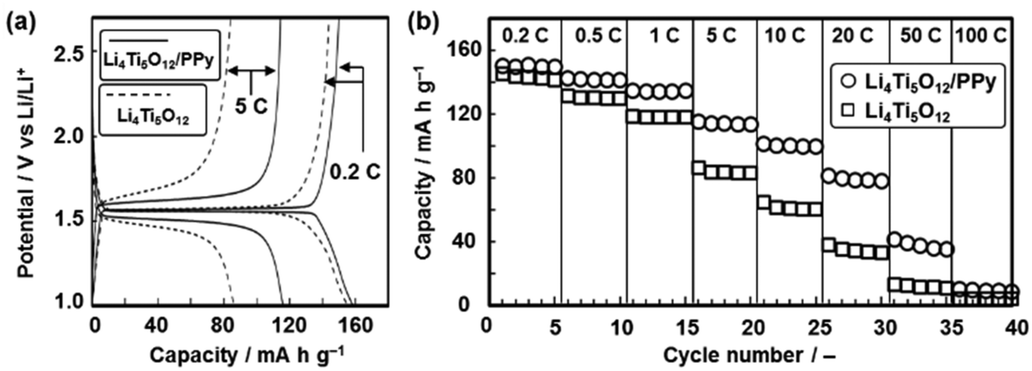 | ||
| Fig. 4 Conductive coating on Li4Ti5O12 nanocrystals, an electrode active material for lithium-ion battery. (a) Charge–discharge curves of the commercial nanocrystals (dashed lines) and the PPy-coated one (solid lines) at the current density of 0.2 C and 5 C (1 C = 0.175 A g−1). (b) Relationship between the current density and the specific capacity of the commercial nanocrystals (squares) and the PPy-coated one (circles). | ||
In summary, conductive polymers consisting of pyrrole and thiophene backbones, such as PPy and PEDOT, have been synthesized and coated on a variety of substrates under ambient pressure and low temperature conditions. The monomers in contact with the oxidant crystals generate the activated monomers. Therefore, the polymerization and coating spontaneously proceed not only on the surface of the oxidant crystals but also on the separately-placed substrates. When the substrate was changed to the hierarchically organized material, the morphology was replicated to the conductive polymer by the casting process. The conductive coating on the nanocrystals of an electrode active material facilitated the improvement of the electrochemical performances. The present approach can be applied to synthesis, coating, and morphogenesis of a variety of conductive polymer materials and other polymers by tuning the synthetic conditions.
This work was partially supported by a Grant-in-Aid for Scientific Research (No. 22107010) on Innovative Areas of “Fusion Materials (No. 2206)” from the MEXT.
Notes and references
- (a) S. Mann, Angew. Chem., Int. Ed., 2000, 39, 3392 CrossRef CAS; (b) M. Antonietti and G. A. Ozin, Chem. – Eur. J., 2004, 10, 28 CrossRef CAS PubMed.
- (a) T. Kato, Adv. Mater., 2000, 12, 1543 CrossRef CAS; (b) S.-H. Yu and H. Cölfen, J. Mater. Chem., 2004, 14, 2124 RSC; (c) F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS PubMed; (d) N. A. J. M. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499 CrossRef CAS PubMed; (e) H. Imai and Y. Oaki, MRS Bull., 2010, 35, 138 CrossRef CAS.
- (a) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38 CrossRef CAS PubMed; (b) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed; (c) J. J. L. M. Comelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039 CrossRef PubMed.
- (a) R. A. Caruso and M. Antonietti, Chem. Mater., 2001, 13, 3272 CrossRef CAS; (b) C. R. Martin, Acc. Chem. Res., 1995, 28, 61 CrossRef CAS.
- (a) H. W. Liang, J. W. Liu, H. S. Qian and S. H. Yu, Acc. Chem. Res., 2013, 46, 1450 CrossRef CAS PubMed; (b) S. J. Hurst, E. M. Payne, L. Qin and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 2672 CrossRef CAS PubMed.
- (a) A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738 CrossRef CAS; (b) T. L. Kelly and M. O. Wolf, Chem. Soc. Rev., 2010, 39, 1526 RSC; (c) K. Sada, M. Takeuchi, N. Fujita, M. Numata and S. Shinkai, Chem. Soc. Rev., 2007, 36, 415 RSC.
- (a) E. Ruiz-Hitzky, Adv. Mater., 1993, 5, 334 CrossRef CAS PubMed; (b) K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950 CrossRef CAS; (c) K. Tajima and T. Aida, Chem. Commun., 2000, 2339 Search PubMed; (d) D. J. Cardin, Adv. Mater., 2002, 14, 553 CrossRef CAS; (e) T. Uemura, S. Horike and S. Kitagawa, Chem. – Asian J., 2006, 12, 36 CrossRef PubMed.
- (a) M. Kijima, Y. Oaki, Y. Munekawa and H. Imai, Chem. – Eur. J., 2013, 19, 2284 CrossRef CAS PubMed; (b) Y. Oaki, M. Kijima and H. Imai, J. Am. Chem. Soc., 2011, 133, 8594 CrossRef CAS PubMed; (c) Y. Munekawa, Y. Oaki and H. Imai, Langmuir, 2014, 30, 3236 CrossRef CAS PubMed.
- H. Sinn and W. Kaminsky, Adv. Organomet. Chem., 1980, 18, 99 CrossRef CAS.
- (a) J. A. Lipton-Duffin, J. A. Miwa, M. Kondratenko, F. Cicoira, B. G. Sumpter, V. Meunier, D. F. Perepichka and F. Rose, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11200 CrossRef CAS PubMed; (b) L. Lafferentx, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215 CrossRef PubMed; (c) K. Onodera, C. Tanioku and A. Matsumoto, ACS Appl. Mater. Interfaces, 2012, 4, 2280 CrossRef CAS PubMed; (d) D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1329 CrossRef CAS PubMed.
- (a) J. Kim, E. Kim, Y. Won, H. Lee and K. Suh, Synth. Met., 2003, 139, 485 CrossRef CAS; (b) J. Kim, M. Kwon, Y. Min, S. Kwon and D. W. Ihm, Adv. Mater., 2007, 19, 3501 CrossRef CAS PubMed; (c) H. Bai, C. Li, F. Chen and G. Shi, Polymer, 2007, 48, 5259 CrossRef CAS PubMed.
- (a) A. Mohammadi, M. A. Hasan, B. Liedberg, I. Lundstrom and W. R. Salaneck, Synth. Met., 1986, 14, 189 CrossRef CAS; (b) D. Bhattacharyya, R. M. Howden, D. C. Borrelli and K. K. Gleason, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1329 CrossRef CAS PubMed; (c) A. M. Coclite, R. M. Howden, D. C. Borrelli, C. D. Petruczok, R. Yang, J. L. Yagüe, A. Ugur, N. Chen, S. Lee, W. J. Jo, A. Liu, X. Wang and K. K. Gleason, Adv. Mater., 2013, 25, 5392 CrossRef CAS PubMed; (d) B. Winther-Jensen and K. West, Macromolecules, 2004, 37, 4538 CrossRef CAS; (e) B. Winther-Jensen, J. Chen, K. West and G. Wallace, Macromolecules, 2004, 37, 5930 CrossRef CAS; (f) S. G. Im, P. J. Yoo, P. T. Hammond and K. K. Gleason, Adv. Mater., 2007, 19, 2863 CrossRef CAS PubMed.
- Y. Yin, Z. Li, J. Jin, C. Tusy and J. Xia, Synth. Met., 2013, 175, 97 CrossRef CAS PubMed.
- (a) Y. Oaki and H. Imai, Small, 2006, 2, 66 CrossRef CAS PubMed; (b) Y. Oaki, A. Kotachi, T. Miura and H. Imai, Adv. Funct. Mater., 2006, 16, 1633 CrossRef CAS PubMed.
- (a) Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294 RSC; (b) G. N. Zhu, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2012, 5, 6652 RSC; (c) H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201 RSC; (d) X. Liu, H. Li, D. Li, M. Ishida and H. Zhou, J. Power Sources, 2013, 243, 373 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Microscopy images, spectroscopic data, and the proposed formation mechanisms. See DOI: 10.1039/c4cc05171h |
| This journal is © The Royal Society of Chemistry 2014 |