
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monotelechelic poly(p-phenylenevinylene)s by ring opening metathesis polymerisation†
Benjamin J.
Lidster
,
Jonathan M.
Behrendt
and
Michael L.
Turner
*
Organic Materials Innovation Centre, School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Michael.Turner@Manchester.ac.uk; Fax: +44-161-275-4273
First published on 18th August 2014
Abstract
Poly(p-phenylenevinylene)s (PPVs) with single reactive end groups have been prepared with high molecular weights, narrow polydispersities (ĐM) and excellent end functionality (f). PPVs functionalised with α-bromoester end groups are effective macroinitiators in the atom transfer radical polymerisation (ATRP) of methyl methacrylate (MMA).
The use of π-conjugated polymers as the active layer in electronic devices has been the subject of intense research over the past 20 years.1 Precise control of both the backbone microstructure and end groups in this important class of polymers is a considerable challenge, principally due to a scarcity of well-controlled routes for their synthesis. Current notable examples include nickel catalysed Grignard metathesis polymerisation to give poly(thiophene)s and palladium catalyst transfer Suzuki–Miyaura polycondensation to give poly(fluorene)s and poly(p-phenylene)s.2 End group control afforded by such routes can lead to improved stability and optimisation of the photophysical and electronic properties, e.g. elimination of reactive groups, minimisation of charge trapping and tuning of the band gap.3 Reactive end groups also provide access to block copolymers via the ‘grafting to’ and ‘grafting from’ methodologies.4 The structure-driven self-assembly of block copolymers (containing one or more π-conjugated segments) holds great promise in organic electronics, where the thin-film morphology of the active material has a dramatic influence on the overall device performance.5
PPVs remain a popular choice of π-conjugated polymer in organic electronic devices due to their favorable electronic and optical properties.6 Synthetic routes to PPVs, while numerous, are usually uncontrolled; giving broad ĐM, poorly defined end groups and non-conjugated defects.7 Controlled polymerisations to PPVs are restricted to precursor routes or limited by the achievable molecular weight.8 The exception is the ROMP of substituted cyclophanedienes and cyclophanetrienes, which provides a direct route to defect-free, structurally defined PPVs of predetermined Mn and narrow ĐM.9 Conjugated diblock copolymers with two different phenylenevinylene segments have also been prepared via this route.10 In an extension of this work, we report the preparation of monotelechelic PPVs bearing α-bromoester and tolyl end groups. The α-bromoester end functionalised PPVs have further been used as macroinitiators in ATRP, giving a series of PPV-b-PMMA copolymers that retain the desirable optical properties (Scheme 1).
 | ||
| Scheme 1 Synthesis of α-bromoester and tolyl functionalised monotelechelic PPVs 4a–d and 5a–d and the corresponding PPV-b-PMMA diblock copolymers 6a–f. | ||
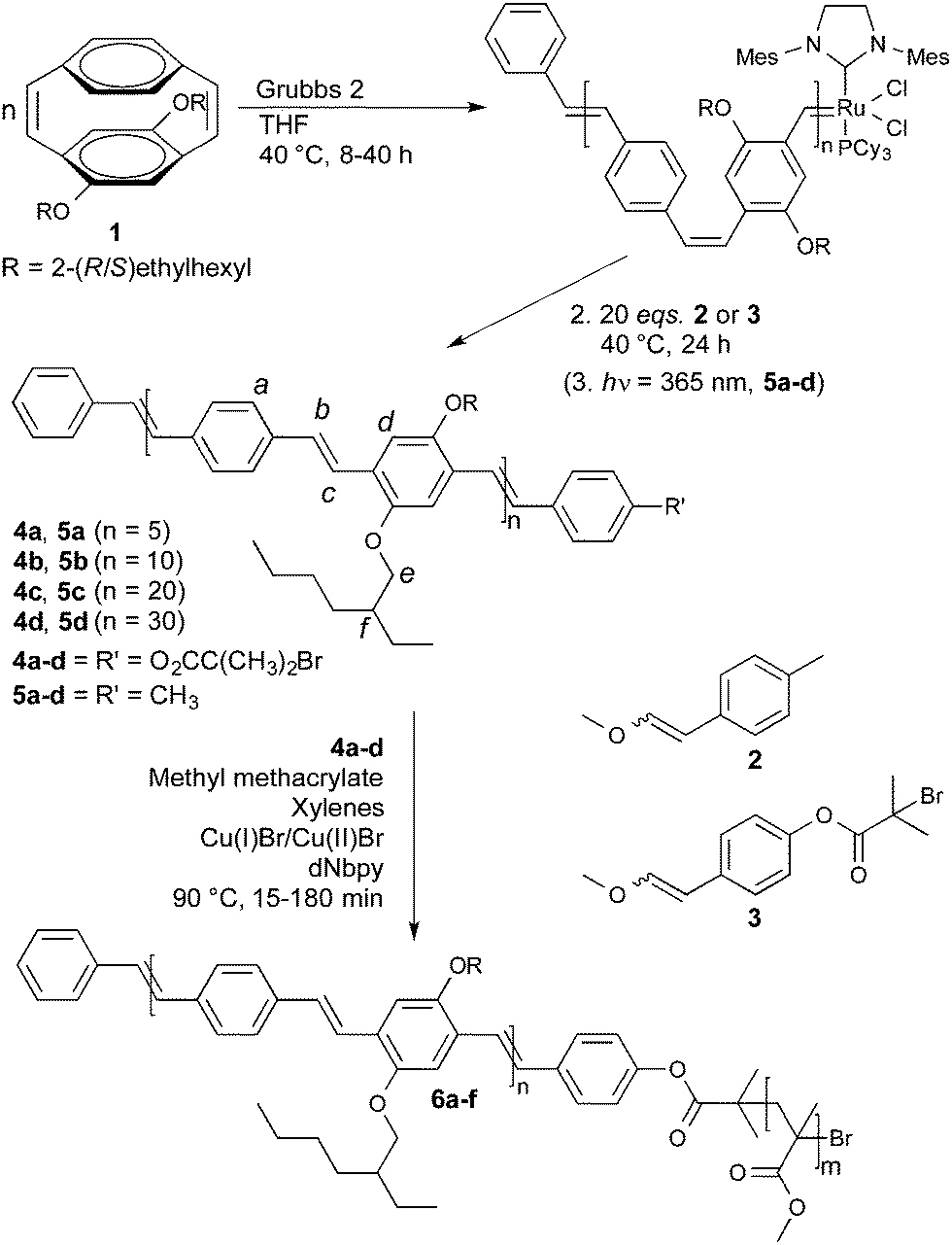
The monomer 4,7-diethylhexyloxy-[2.2]paracyclophane-1,9-diene (1) was prepared by a modification of the previously reported procedure.11 The ROMP of cyclophanediene 1 was initiated using the Grubbs 2 complex in THF at 40 °C. The number average degree of polymerisation (xn) was effectively controlled by changing the [1]/[Grubbs 2] ratio, as expected for a living polymerisation. In previous reports, quenching of the active ruthenium carbene chain end with an excess of ethyl vinyl ether resulted in each PPV chain containing a vinyl and a phenyl end group.9a Monotelechelic PPVs with one functional end group should therefore be prepared by quenching the polymerisation with vinyl ethers carrying the desired functionality. The ROMP of cyclophanediene 1 is efficiently quenched on addition of 20 eq. of either α-bromoester functionalised vinyl ether 2 or tolyl functionalised vinyl ether 3, at 40 °C for 24 hours. α-Bromoester functionalised polymers 4a–d and the corresponding tolyl terminated homopolymers 5a–d were isolated as orange films in excellent yields (83–90%), after purification by multiple precipitations onto a short methanol/Celite column and extraction with hot chloroform. 1H NMR spectroscopy of polymer 4b confirmed the incorporation of the α-bromoester group with a sharp singlet at 2.09 ppm corresponding to the two CH3 groups of the ester (Fig. 1(a)). The OCH2 of the main polymer chain are observed at 3.49 and 3.98 ppm, corresponding to adjacent vinylene bonds in the Z and E stereochemistry, respectively (other key signals are assigned in Fig. 1(a)).12 The end group functionalisation (f) was calculated by integration of the two CH3 groups of the terminal α-bromoester group against H–f. For example in polymer 4b the expected integrals from the ratio of [1]/[Grubbs 2] are 6.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20.0 and the observed integrals are 6.0:20.7, indicating a value for f of 97%. All values of f are above 90% for polymers 4a–d and above 85% for polymers 5a–d (Table 1). Typically in ROMP f is determined by integration of key signals associated with each end group, however in this instance signals for the phenyl end group (derived from the Grubbs 2 initiating species) are obscured due to overlap with signals of the polymer backbone.13 Only one vinylene bond of cyclophanediene 1 is subject to metathesis, so the expected polymer backbone microstructure should consist of alternating E/Z-vinylene bonds.9a Surprisingly, and conveniently, polymers 4a–d were isolated with the all E-vinylene stereochemistry (97% as determined by 1H NMR spectroscopy), probably due to isomerisation during work-up. Polymers 5a–d were obtained in the all E-vinylene stereochemistry after photoisomerisation.10
:20.0 and the observed integrals are 6.0:20.7, indicating a value for f of 97%. All values of f are above 90% for polymers 4a–d and above 85% for polymers 5a–d (Table 1). Typically in ROMP f is determined by integration of key signals associated with each end group, however in this instance signals for the phenyl end group (derived from the Grubbs 2 initiating species) are obscured due to overlap with signals of the polymer backbone.13 Only one vinylene bond of cyclophanediene 1 is subject to metathesis, so the expected polymer backbone microstructure should consist of alternating E/Z-vinylene bonds.9a Surprisingly, and conveniently, polymers 4a–d were isolated with the all E-vinylene stereochemistry (97% as determined by 1H NMR spectroscopy), probably due to isomerisation during work-up. Polymers 5a–d were obtained in the all E-vinylene stereochemistry after photoisomerisation.10
Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF-MS) of the isolated polymer 4b exhibited one major series corresponding to polymer chains with both α-bromoester and phenyl end groups (●), with the major series of peaks separated by the mass of the polymer repeat unit (461 Da) (Fig. 1(b)). Two additional minor series were observed; corresponding to polymers with a phenol end group, derived from the cleavage of the terminal ester bond (■) and a vinyl end group by elimination of hydrogen bromide (○).14 The relative intensity of these two series was observed to increase with increasing laser power, suggesting that they result from fragmentation during the MALDI experiment (ESI†). For polymer 4b no species were observed with a carbonyl end group, resulting from the decomposition of the ruthenium carbene chain end or polymers with a methyl vinyl ether end group. Species resulting from secondary metathesis were observed to occur in low abundance, as has previously reported.10
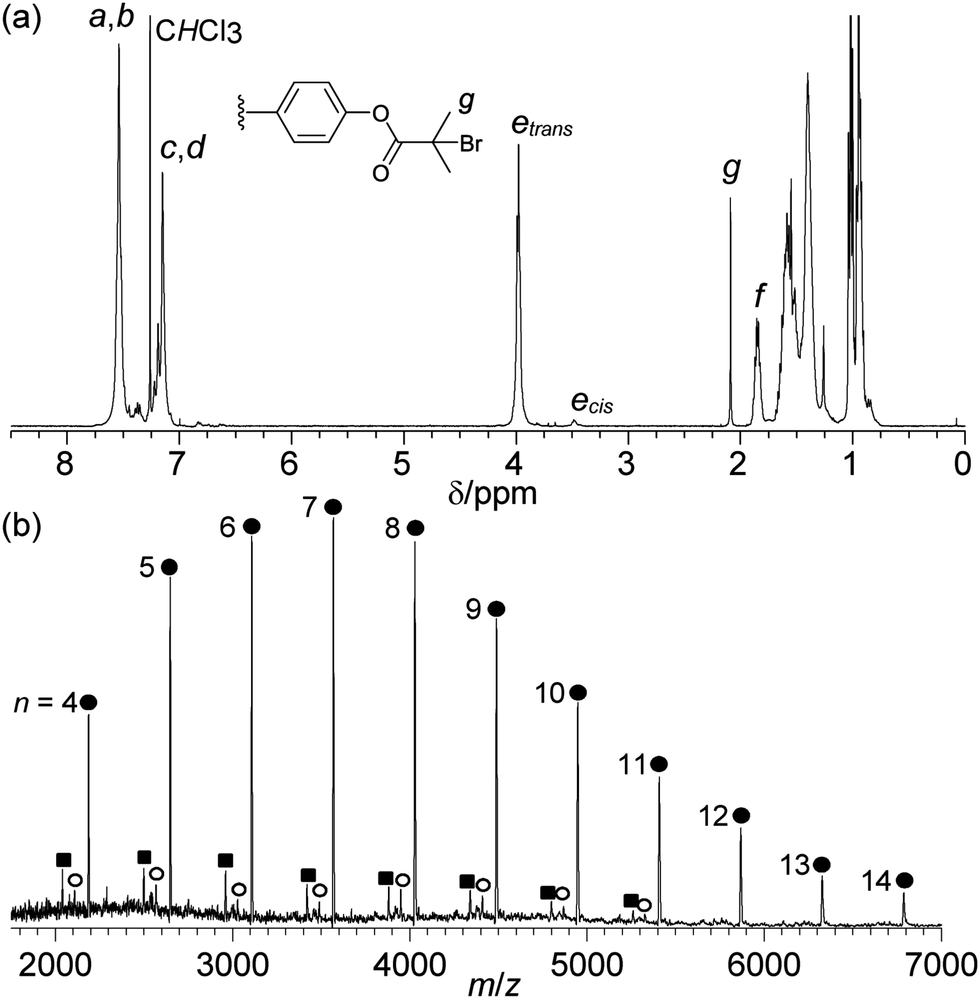 | ||
| Fig. 1 (a) 1H NMR spectrum of polymer 4b, (b) MALDI-TOF-mass spectrum of polymer 4b (xn indicated). | ||
| Polymer | x n (PPV) | M n (kg mol−1) | x m (PMMA) | M n (kg mol−1) | Đ M | f (%) | λ max (nm) | λ em (nm) | PLQYd |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated from the [1]/[Grubbs 2] ratio, inc. expected end groups. b Determined by 1H NMR spectroscopy. c Determined by GPC with RI detection (calibrated against narrow ĐM polystyrene standards). d λ Ex = 470 nm, against fluorescein standards (0.1 M sodium hydroxide(aq)), in DCM. | |||||||||
| 4a | 5 | 2.6 | — | 4.7c | 1.36 | 92 | — | — | — |
| 4b | 10 | 5.0 | — | 9.0c | 1.51 | 97 | — | — | — |
| 4c | 20 | 9.6 | — | 22.8c | 1.44 | 100 | — | — | — |
| 4d | 30 | 14.1 | — | 39.0c | 1.45 | 93 | — | — | — |
| 5a | 5 | 2.5 | — | 3.7c | 1.50 | 95 | 470 | 525 | 0.58 |
| 5b | 10 | 4.8 | — | 8.8c | 1.51 | 94 | 476 | 526 | 0.64 |
| 5c | 20 | 9.4 | — | 18.8c | 1.47 | 85 | 482 | 527 | 0.65 |
| 5d | 30 | 14.0 | — | 37.4c | 1.30 | 88 | 484 | 528 | 0.65 |
| 6a | 5 | — | 800 | 82.7b | 1.19 | — | 466 | 525 | 0.59 |
| 6b | 10 | — | 750 | 79.9b | 1.33 | — | 476 | 528 | 0.58 |
| 6c | 20 | — | 450 | 54.5b | 1.32 | — | 481 | 529 | 0.63 |
| 6d | 20 | — | 800 | 87.6b | 1.28 | — | 483 | 529 | 0.64 |
| 6e | 20 | — | 1350 | 143.6b | 1.31 | — | 483 | 529 | 0.67 |
| 6f | 30 | — | 800 | 90.1b | 1.31 | — | 486 | 529 | 0.66 |
Gel permeation chromatography (GPC) analysis of polymers 4a–d and 5a–d exhibited a linear correlation between the Mn and the [1]/[Grubbs 2] ratio, indicating no loss of control during the polymerisation or importantly during the termination reaction (ESI†). Polymers 4a–d were isolated with unimodal distributions and ĐM in the range of 1.36–1.51. As polymers 4a–d were isolated predominantly in the all E-vinylene stereochemistry, an increase in the hydrodynamic volume of the chains over that of a simple random coil is observed. The apparent Mn values are approximately two times greater than that of the predicted Mn, when measured against the polystyrene calibration standards.
The controlled radical polymerisation of activated vinyl monomers using ATRP gives well-defined polymers (e.g. PMMA), that are not easily accessible using ROMP.15 Block copolymers can be prepared using this polymerisation technique by the use of a suitably functionalised macroinitiators. Polymers 4a–d were found to be effective macroinitiators for ATRP of MMA, resulting in PPV-b-PMMA block copolymers (6a–f) with excellent control of both segments. Although polymers 4a–d are readily soluble in dilute solutions of solvents such as THF, CHCl3, CH2Cl2, they are not fully soluble at the concentrations typically used in ATRP. Hence the synthesis of the diblock copolymers was performed with concentration of polymers 4a–d of between 1.7–11.1 mM, using a solution of [MMA]:[xylenes]:[Cu(I)Br]:[Cu(II)Br2]:[dNbpy] = [800]:[800]:[1]:[0.05]:[2] at 40 °C. The polymerisation was initiated by heating to 90 °C, with longer reaction times required to obtain polymers with higher molecular weight PMMA segments.
ATRP was terminated by exposure to air and the polymeric products purified by precipitation into methanol, followed by reprecipitation into diethyl ether from chloroform. The desired diblock copolymers 6a–f were isolated as bright orange powders.
The 1H NMR spectrum of diblock copolymer 6c obtained from the corresponding macroinitiator 4c, (Fig. 2(a)) clearly displays peaks associated with both the PPV and PMMA segments. 1H NMR spectroscopy was used to determine the xn of the PMMA segment by integration of the OCH3 of PMMA against the OCH2 of the PPV segment. For the diblock copolymer 6c a xm = 450 for the PMMA segment was calculated, resulting in an assigned structure of PPV20-b-PMMA450.
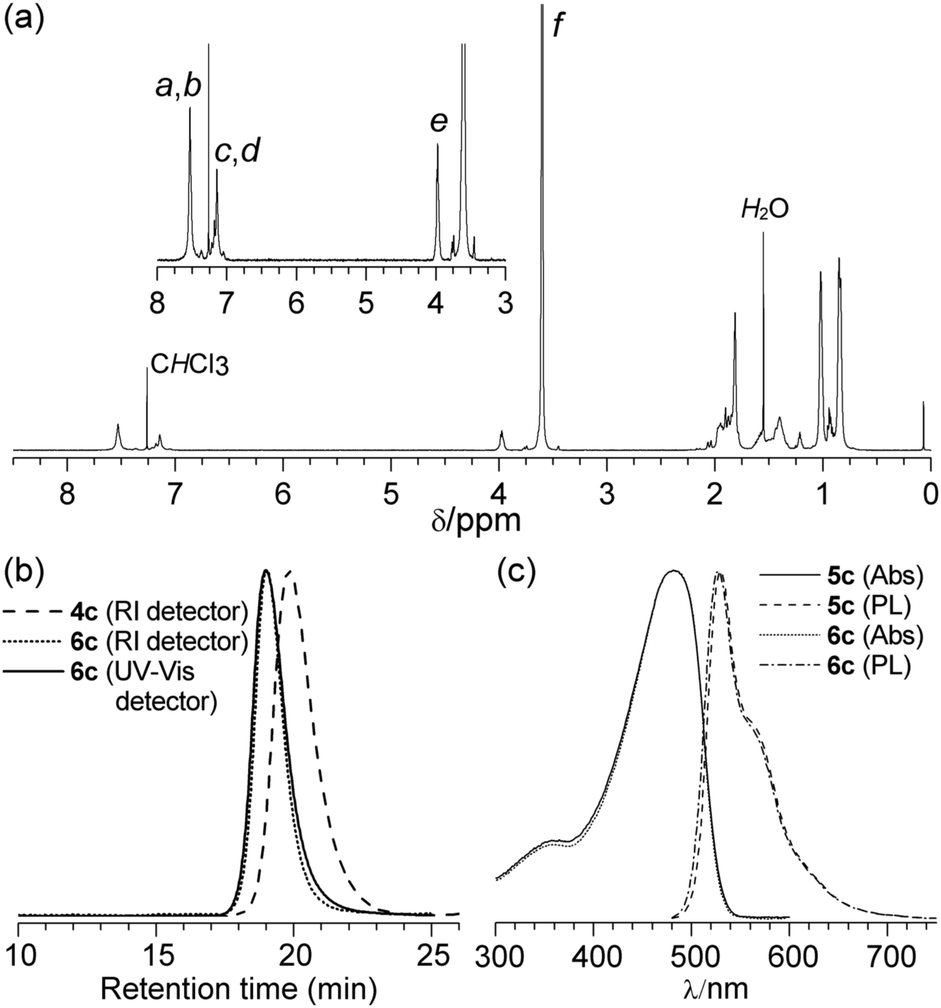 | ||
| Fig. 2 (a) 1H NMR spectrum of polymer 6c, (b) GPC chromatograms of polymer 4c and diblock copolymer 6c, (c) UV-Vis absorption and PL spectra of diblock copolymer 6c and the corresponding homopolymer 5c. | ||
Representative GPC chromatograms for macroinitiator 4c and the corresponding diblock copolymer 6c are shown in Fig. 2(b). The ĐM was observed to decrease from 1.44 for macroinitiator 4c to 1.32 for polymer 6c, with an apparent Mn = 46.0 kg mol−1 and is consistent with a controlled chain growth polymerisation. As PPV is strongly absorbing in the ultraviolet-visible region and PMMA is not, use of GPC equipped with both a UV-Vis detector (set at 450 nm) and a refractive index (RI) detector allowed for the detection of any residual macroinitiator 4c and for possible side reactions during the ATRP. The GPC trace for block copolymer 6c (Fig. 2(b)) shows no residual macroinitiator 4c and concurrent unimodal distributions on both the RI and UV-Vis detectors. The absence of macroinitiator 4c indicates a high degree of initiation in addition to the selective precipitation of the diblock copolymer 6c. No evidence for termination by radical–radical coupling was observed, which would result in PPV-b-PMMA-b-PPV triblock copolymers. Slight tailing on the low molecular weight side, in particular to the UV-Vis chromatogram can be attributed to a small contribution from competing termination reactions, which are commonly observed in ATRP. The discrepancy between the apparent Mn from GPC and the Mn calculated from 1H NMR spectroscopy is due to the hydrodynamic volume of the diblock copolymer 6c being dominated by the rigid PPV segment.
The influence of the relative block lengths of the PPV-b-PMMA copolymers on the solution-phase optical properties, was studied by preparation of a series of polymers, 6a–f, with varying PPV (n) and PMMA (m) block lengths. The UV-Vis and photoluminescence (PL) spectra and photoluminescence quantum yields (PLQY) were measured (molecular weight data in Table 1, n values were determined from [1]/[Grubbs 2] ratio, m values are rounded to the nearest 50 for ease of comparison). As expected, a red shift in the λmax of the polymers was observed on increasing the conjugation length of the PPV block.
The length of the PMMA block has no discernible influence on the position of the maximum absorption (Table 1). Only a slight increase in the PLQY with increasing xn of the PPV segment was observed and no significant variation with increasing xn of the PMMA segment. For comparison, a series of polymers 5a–d with similar PPV block lengths to each of the block-copolymers were also studied. Fig. 2(c) shows the UV-Vis and PL spectra of block copolymer 6c and homopolymer PPV20. It highlights the very close similarity of the absorption and emission profiles of the block copolymer and the corresponding homopolymer. Furthermore, there were no significant differences in PLQY between the diblock copolymers and homopolymers, providing further evidence that the PPV conjugated backbone is not degraded by the ATRP reaction conditions.
In summary high molecular weight poly(p-phenylenevinylene-2,5-diethylhexyloxy-p-phenylenevinylene)s with functional end groups can be prepared by the termination of a living ROMP of cyclophanedienes with suitably functionalised vinyl ethers. Excellent control of the molecular weight, narrow ĐM and a high f are possible using this route. Functionalisation with α-bromoester groups results in PPVs which are effective macroinitiators for the ATRP of methyl methacrylate. PPV-b-PMMA diblock copolymers were obtained with unimodal distributions, narrow ĐM, which are free from any residual PPV homopolymer. The solution-phase self-assembly and solid-state morphologies of these diblock copolymers are currently under investigation.
We thank the EPSRC and JSPS for financial support for BL, the TSB for support for OMIC and Gareth Smith for help with MALDI-TOF mass spectrometry.
Notes and references
- (a) J. B. You, L. T. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed; (b) N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474 CAS; (c) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS PubMed.
- (a) E. Elmalem, F. Biedermann, K. Johnson, R. H. Friend and W. T. S. Huck, J. Am. Chem. Soc., 2012, 134, 17769 CrossRef CAS PubMed; (b) D. Marsitzky, M. Klapper and K. Müllen, Macromolecules, 1999, 32, 8685 CrossRef CAS; (c) M. C. Stefan, M. P. Bhatt, P. Sista and H. D. Magurudeniya, Polym. Chem., 2012, 3, 1693 RSC; (d) C. S. Fischer, M. C. Baier and S. Mecking, J. Am. Chem. Soc., 2012, 135, 1148 CrossRef PubMed; (e) M. Jeffries-El, G. Sauvé and R. D. McCullough, Macromolecules, 2005, 38, 10346 CrossRef CAS.
- (a) M. J. Robb, D. Montarnal, N. D. Eisenmenger, S.-Y. Ku, M. L. Chabinyc and C. J. Hawker, Macromolecules, 2013, 46, 6431 CrossRef CAS; (b) Q. Wang, B. Zhang, L. Liu, Y. Chen, Y. Qu, X. Zhang, J. Yang, Z. Xie, Y. Geng, L. Wang and F. Wang, J. Phys. Chem. C, 2012, 116, 21727 CrossRef CAS; (c) Z. Mao, K. Vakhshouri, C. Jaye, D. A. Fischer, R. Fernando, D. M. DeLongchamp, E. D. Gomez and G. Sauvé, Macromolecules, 2012, 46, 103 CrossRef; (d) Y. Kim, S. Cook, J. Kirkpatrick, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, M. Heeney, R. Hamilton and I. McCulloch, J. Phys. Chem. C, 2007, 111, 8137 CrossRef CAS.
- M. Hillmyer, Curr. Opin. Solid State Mater. Sci., 1999, 4, 559 CrossRef CAS.
- (a) A. Yassar, L. Miozzo, R. Gironda and G. Horowitz, Prog. Polym. Sci., 2013, 38, 791 CrossRef CAS PubMed; (b) H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217 CrossRef CAS; (c) U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS; (d) X. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579 CrossRef CAS PubMed; (e) H. Hoppe and N. S. Sariciftci, J. Mater. Chem., 2006, 16, 45 RSC; (f) R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205 CrossRef CAS; (g) T.-Q. Nguyen, I. B. Martini, J. Liu and B. J. Schwartz, J. Phys. Chem. B, 1999, 104, 237 CrossRef.
- (a) W. Wang, J. H. Alsmeier and R. Schlaf, Langmuir, 2013, 29, 6341 CrossRef CAS PubMed; (b) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS; (c) A. Köhler, S. T. Hoffmann and H. Bässler, J. Am. Chem. Soc., 2012, 134, 11594 CrossRef PubMed.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef.
- (a) H. Kretzschmann and H. Meier, Tetrahedron Lett., 1991, 32, 5059 CrossRef CAS; (b) I. Cosemans, J. Vandenbergh, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2013, 4, 3471 RSC.
- (a) C.-Y. Yu and M. L. Turner, Angew. Chem., Int. Ed., 2006, 45, 7797 CrossRef CAS PubMed; (b) U. H. F. Bunz, D. Mäker and M. Porz, Macromol. Rapid Commun., 2012, 33, 886 CrossRef CAS PubMed; (c) D. Mäker, C. Maier, K. Brödner and U. H. F. Bunz, ACS Macro Lett., 2014, 415 CrossRef.
- C.-Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2010, 43, 222 CrossRef CAS.
- C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem. – Eur. J., 2011, 17, 6991 CrossRef CAS PubMed.
- H. Katayama, M. Nagao, T. Nishimura, Y. Matsui, Y. Fukuse, M. Wakioka and F. Ozawa, Macromolecules, 2006, 39, 2039 CrossRef CAS.
- E. J. Gordon, J. E. Gestwicki, L. E. Strong and L. L. Kiessling, Chem. Biol., 2000, 7, 9 CrossRef CAS.
- A. Can, E. Altuntas, R. Hoogenboom and U. S. Schubert, Eur. Polym. J., 2010, 46, 1932 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of cyclophanediene 1, vinyl ether 3, polymers 4a–d, 5a–d and 6a–f. MALDI-TOF-MS of polymer 5b, molecular weight distribution of polymers 4a–d, 5a–d, and GPC chromatograms of block copolymers 6a–f. See DOI: 10.1039/c4cc05118a |
| This journal is © The Royal Society of Chemistry 2014 |