
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous synthesis of artemisinin-derived medicines†
Kerry
Gilmore
 a,
Daniel
Kopetzki
a,
Ju Weon
Lee
b,
Zoltán
Horváth
b,
D. Tyler
McQuade
a,
Andreas
Seidel-Morgenstern
bc and
Peter H.
Seeberger
*ad
a,
Daniel
Kopetzki
a,
Ju Weon
Lee
b,
Zoltán
Horváth
b,
D. Tyler
McQuade
a,
Andreas
Seidel-Morgenstern
bc and
Peter H.
Seeberger
*ad
aMax-Planck-Institute of Colloids and Interfaces, Department of Biomolecular Systems, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: peter.seeberger@mpikg.mpg.de
bMax-Planck-Institute for Dynamics of Complex Technical Systems, Sandtorstrasse 1, 39106 Magdeburg, Germany
cOtto-von-Guericke-University, Chair for Chemical Process Technology, Universitätsplatz 2, 39106 Magdeburg, Germany
dFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany
First published on 29th August 2014
Abstract
Described is a continuous, divergent synthesis system which is coupled to continuous purification and is capable of producing four anti-malarial APIs. The system is comprised of three linked reaction modules for photooxidation/cyclization, reduction, and derivatization. A fourth module couples the crude reaction stream with continuous purification to yield pure API.
As the global demand for medicines has increased, efforts to implement continuous Active-Pharmaceutical Ingredient (API) production have intensified.1 Continuous processes hold many potential benefits including increased safety and product quality, as well as lower environmental impact.2 While proof-of-concept continuous API syntheses3,4 focused on linear transformations, we describe the combination of a divergent, multi-step continuous synthesis with continuous in-line purification to prepare several artemisinin-derived APIs that are key components in Artemisinin Combination Therapies (ACT), the World Health Organization's (WHO) recommended first-line treatment for malaria.
Today, the key active pharmaceutical ingredients of all ACT anti-malarials are produced in one or two chemical steps from artemisinin (3) (Fig. 1). The majority of artemisinin (∼200 tons per year) is extracted from the plant Artemisia annua and prices fluctuate with harvest yields.5 The plant waste material dihydroartemisinic acid (DHAA) 2 has also been used to synthesise artemisinin via continuous flow photochemistry with subsequent acid-induced rearrangement, resulting in a fast (residence time less than 12 minutes) and efficient (69% yield) transformation.6,7
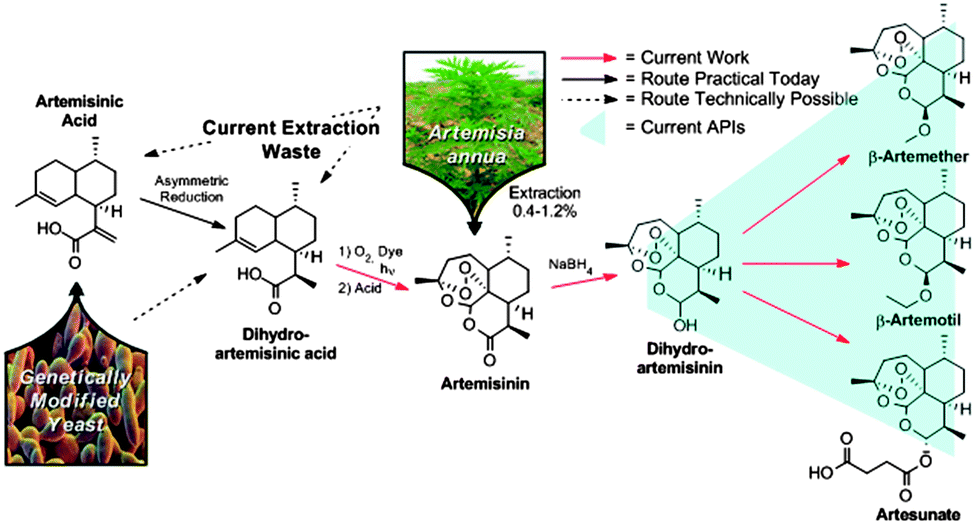 | ||
| Fig. 1 Overall production scheme of anti-malaria APIs from artemisinin obtained by traditional extraction from Artemisia annua and genetically modified yeast combined with chemical modification. Dihydroartemisinin (4, combined with piperaquine in Eurartesim, Artekin and Duo-Cotecxin), β-artemether (5, combined with lumefantrine in Coartem), β-arteether (6, Artemotil), and α-artesunate (7, combined with amodiaquine in Coarsucam and ASAQ-Winthrop). | ||
ACT medications rely not only on the procurement of artemisinin, but also its conversion to the corresponding APIs (4–7). To achieve a completely continuous synthesis and purification of the three main anti-malarial APIs, we built upon the previously described continuous synthesis of artemisinin 3 from DHAA 2 (Fig. 3, module 1).6 While lithium triethylborohydride (Super-hydride®) effects the reduction of pure artemisinin 3 to DHA 4 in a continuous flow chemistry setting,8 this reagent is an order of magnitude more expensive than the NaBH4 used in traditional batch syntheses. Super-hydride® is even more expensive than artemisinin, making it unsuitable for the production of a price-sensitive API. Consequently, a continuous process using inexpensive NaBH4 had to be developed.9
Several challenges had to be overcome, including the insolubility of NaBH4 in the toluene–TFA solvent mixtures exiting module 1. While NaBH4 suspensions have been used in batch to prepare DHA, the use of slurries in flow remains a major challenge,10 particularly when envisioning prolonged production runs. A NaBH4-containing packed-bed column would be an attractive means of reducing 3 to 4. While packed-beds containing NaBH4/Celite® mixtures (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w))11 successfully reduced artemisinin from the outflow of module 1 to DHA, columns packed in this way proved unstable (see ESI†). Different column additives to both accelerate the reduction and stabilise the column were screened (Fig. 2). Lithium chloride (LiCl) accelerates NaBH4 reductions via the in situ formation of LiBH4,12 but clogging remained a problem. Buffering the trifluoroacetic acid (TFA) contained in the reaction stream by the addition of lithium carbonate (Li2CO3) also did not remedy the clogging problems. However, upon combining the two additives and mixing NaBH4, Celite®, Li2CO3, and LiCl (1:1:1:0.76 (w/w)) as packed-bed materials and using an alcohol co-solvent such as ethanol,12 complete and clean reduction of crude artemisinin was achieved (Fig. 2, red line). By combining modules 1 and 2, the first API (4) is produced from DHAA 2, with module 2 representing a general solution to achieve efficient reductions of carbonyl compounds in continuous flow, with a reduction capacity which is stoichiometric with respect to NaBH4.13
:1 (w/w))11 successfully reduced artemisinin from the outflow of module 1 to DHA, columns packed in this way proved unstable (see ESI†). Different column additives to both accelerate the reduction and stabilise the column were screened (Fig. 2). Lithium chloride (LiCl) accelerates NaBH4 reductions via the in situ formation of LiBH4,12 but clogging remained a problem. Buffering the trifluoroacetic acid (TFA) contained in the reaction stream by the addition of lithium carbonate (Li2CO3) also did not remedy the clogging problems. However, upon combining the two additives and mixing NaBH4, Celite®, Li2CO3, and LiCl (1:1:1:0.76 (w/w)) as packed-bed materials and using an alcohol co-solvent such as ethanol,12 complete and clean reduction of crude artemisinin was achieved (Fig. 2, red line). By combining modules 1 and 2, the first API (4) is produced from DHAA 2, with module 2 representing a general solution to achieve efficient reductions of carbonyl compounds in continuous flow, with a reduction capacity which is stoichiometric with respect to NaBH4.13
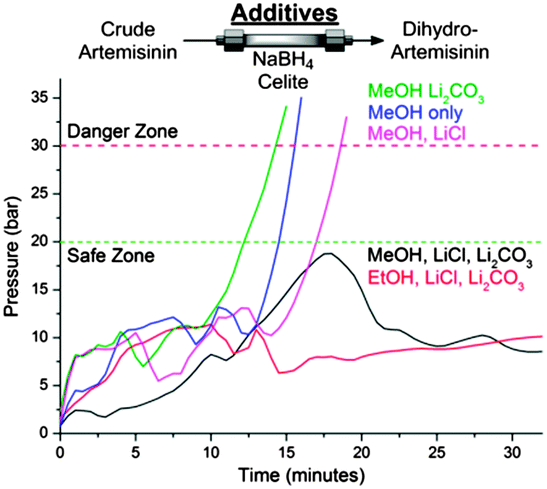 | ||
| Fig. 2 System pressure observed during reduction of crude artemisinin 3 as a function of cosolvent and solid additives in the Celite®/NaBH4 column. | ||
The mixed methyl acetal 5, API of the drug Coartem, is accessed by combining artemisinin reduction and methyl ether formation (modules 2 and 3, Fig. 3). Previous attempts at developing a combined continuous process were unsuccessful due to sensitivity following NaBH4 reduction.14 As such, the effects of both hydride source (1 M LiEt3BH or reduction column) and workup conditions on the resulting epimeric ratio of β:α artemether were examined in batch following treatment with Feed BAM (1.8 M HCl in 2:1 v/v methanol/trimethyl orthoformate). The ratio was determined by 1H NMR of the crude reaction mixture following basic workup of the second step, comparing the doublets at 4.65 ppm (β-artemether 5, J = 4 Hz) and 4.32 ppm (α-artemether, J = 8 Hz).15
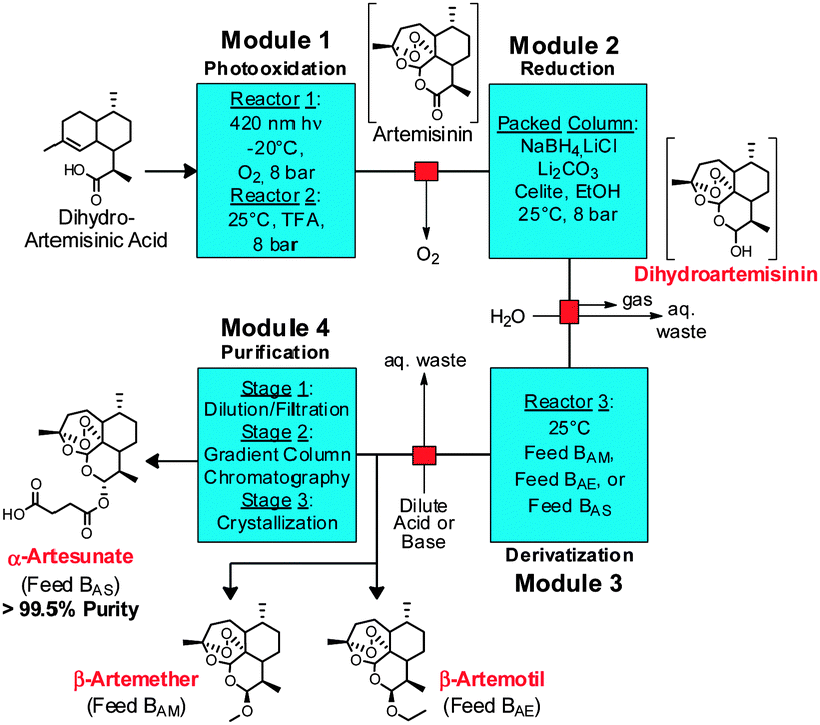 | ||
| Fig. 3 Four module system for the continuous synthesis and purification of artemisinin APIs (in red text). TFA: trifluoroacetic acid, EtOH: ethanol. | ||
As expected, the telescoping of the reduction and mixed acetal formation without any intermediate workup resulted in a poor mixture of artemether epimers for both hydride sources (Table 1, entries 1 and 4).14 It appears the intermediate alkoxyborane species that is formed upon reduction using LiEt3BH is stronger than that emanating from Na/LiBH4, thus requiring an ethanolamine wash to break it down and give acceptable epimeric ratios (entries 2 and 3). However, upon exiting the reduction column, a simple aqueous wash (entry 5) was sufficient to obtain epimeric ratios equal to those resulting from pure dihydroartemisinin.14
| Entry | Hydride source | Intermediate wash | β:α |
|---|---|---|---|
| 1 | Superhydride® (LiHEt3) | None | 50:50 |
| 2 | Superhydride® (LiHEt3) | H2O | 50:50 |
| 3 | Superhydride® (LiHEt3) | H2O/ethanolamine (3/1, v/v) | 80:20 |
| 4 | NaBH4 Column | None | 75:25 |
| 5 | NaBH4 Column | H2O | 81:19 |
| 6 | NaBH4 Column | H2O/ethanolamine (3/1, v/v) | 82:18 |
In flow, the reaction stream exiting module 2 (pH ∼ 10) was continuously degassed16 and extracted17 with water before the organic stream was combined with Feed BAMvia a T-mixer. Complete etherification occurred within a 25 minute residence time at 25 °C, providing artemether 5 as a 81:19 ratio of β:α epimers, similar to that obtained via traditional batch synthesis.14 β-Artemether 5 was obtained in 25% overall yield from DHAA 2 following basic workup and isolation. In a similar manner, artemotil 6 was obtained as a 5:1 mixture of epimers (β:α) in 22% yield from 2 when Feed BAE consisted of a 1.8 M HCl solution in ethanol/triethyl orthoformate (2:1 v/v).
Artesunate 7, the active ingredient in Coarsucam, was continuously prepared from DHAA using a succinic anhydride solution (1.6 M containing one equiv. triethylamine in dichloromethane) in Feed BAS at a 25 minute residence time at 25 °C. Exclusively the desired α-epimer was formed, to yield 7 in 28% from DHAA 2. The crude solution of this derivative was used to demonstrate continuous purification (vide infra).
The entire three module continuous flow process to convert 2 to the APIs 5–7 requires a reaction time of 46 minutes (3.4 mmol per hour through the laboratory-scale system). Importantly, not only can 5, 6, or 7 be synthesised simply by exchanging the solution in Feed B and the acid/base quench (Fig. 3), any further artemisinin C10 derivatives that are under investigation as future anti-malarial APIs18 can be prepared by using this modular synthesis system.
With the chemical steps defined, continuous monitoring and purification were introduced. FlowIR has been shown to be efficient to observe continuous systems in real time19 and was utilized to monitor the system after each reaction module. In module 1, the plant waste material DHAA 2 is converted to artemisinin 3. When the lamp was turned off to simulate a “lamp failure”, the result is observed approximately 11 minutes later when product 3 (1033 cm−1) was no longer observed in the IR trace (Fig. 4). Similarly, the outputs from modules 2 and 3 were also successfully monitored (see ESI†), allowing for facile quality control at each stage of production.
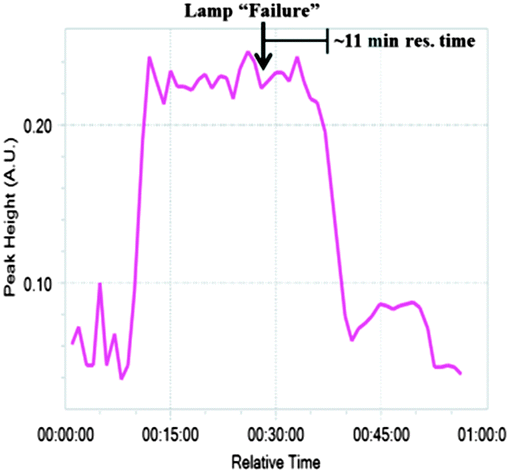 | ||
| Fig. 4 The reaction exiting module 1 was monitored (FlowIR) at 1033 cm−1. To simulate an equipment error, the lamp was turned off during the reaction, resulting in cessation of the monitored peak following the ∼11 minute residence time of module 1. | ||
The continuous synthesis of drug substances is ideally complemented by continuous purification to yield pure APIs that meet the standards set by regulatory bodies such as the FDA20 or WHO.21 To demonstrate the power of the fully continuous synthesis/purification regime, a continuous three-stage purification method (filtration/multi-column chromatography/crystallisation) was developed for α-artesunate 7. The first stage dilutes the stream exiting module 3 with n-hexane, which decreases the solvent strength and precipitates polar byproducts. The recovery yield of 7 after dilution and filtration of the reaction mixture is 65%. Following filtration, the artesunate-containing stream can enter a separator consisting of five identical chromatography columns participating in the following steps: loading, elution, rinsing, and equilibration (see ESI†). This multi-column arrangement, together with the periodic switching between two filters, permits a permanent feed supply. The artesunate fraction can be subsequently fed directly into a continuous crystalliser to remove the remaining impurities (Fig. 5). With 73% of 7 recovered from the crystalliser, the overall recovery yield of α-artesunate from the crude exit stream of module 3 is 48% with an HPLC purity of greater than 99.5%, exceeding the requirements set by the WHO.21
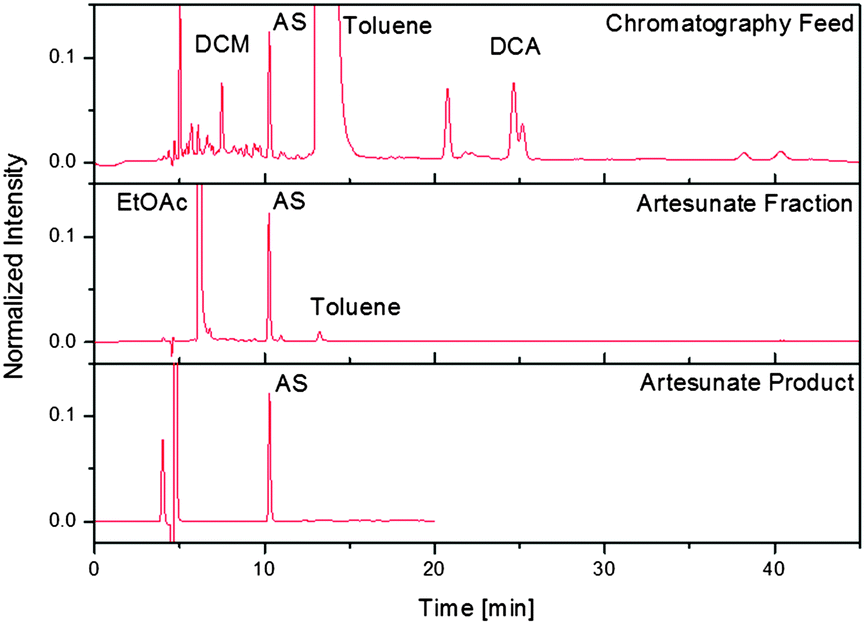 | ||
| Fig. 5 Comparison of HPLC chromatograms exiting module 3 (top), following gradient elution chromatography (middle), and recrystallisation (bottom). Initial peaks in the bottom frame correspond to ethanol injection marks and the run was stopped after 20 minutes due to lack of peaks observed in the chromatogram following the previous purification step (middle). DCM: dichloromethane, EtOAc: ethyl acetate, AS: α-artesunate, DCA: 9,10-dicyanoanthracene. | ||
This continuous, modular approach for the divergent synthesis of small molecules has attractive scientific and conceptual implications. Batch processes are significantly more labour-intensive, as they require solvent removal and purification of the intermediates following each step. The use of potentially dangerous reagents, such as oxygen or sodium borohydride, becomes possible in flow and allows for new reaction pathways,22 and the ability to combine/interchange different reaction modules allows for the production of a variety of APIs. The synthesis of the four anti-malarial APIs described here illustrates this principle and with this first example of a divergent, multi-step continuous synthesis and purification process, many routes to APIs of generic and patented drugs can be envisioned.
We gratefully acknowledge financial support from the Max-Planck Society. We thank Mettler-Toledo for the use of the FlowIR instrument.
Notes and references
- (a) S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans and T. F. Jamison, et al. , Angew. Chem., Int. Ed., 2013, 52, 12359 CrossRef CAS PubMed; (b) P. L. Heider, S. C. Born, S. Basak, B. Benyahia, R. Lakerveld and H. Zhang, et al. , Org. Process Res. Dev., 2014, 18, 402 CrossRef CAS.
- I. W. Davies and C. J. Welch, Science, 2009, 325, 701 CrossRef CAS PubMed.
- (a) M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450 RSC; (b) J. Hartwig, S. Ceylan, L. Kupracz, L. Coutable and A. Kirschning, Angew. Chem., Int. Ed., 2013, 52, 9813 CrossRef CAS PubMed; (c) A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547 CrossRef CAS PubMed; (d) M. Viviano, T. N. Glasnov, B. Reichart, G. Tekautz and C. O. Kappe, Org. Process Res. Dev., 2011, 15, 858 CrossRef CAS; (e) D. R. Snead and T. F. Jamison, Chem. Sci., 2013, 4, 2822 RSC; (f) L. Kupracz and A. Kirschning, Adv. Synth. Catal., 2013, 355, 3375 CrossRef CAS PubMed; (g) M. D. Hopkin, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2013, 11, 1822 RSC.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849 RSC.
- R. Van Noorden, Nature, 2010, 466, 672 CrossRef CAS PubMed.
- (a) D. Kopetzki, F. Lévesque and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 5450 CrossRef CAS PubMed . For our initial work using a medium-pressure Hg lamp, see: ; (b) F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706 CrossRef PubMed.
- This process can be combined with continuous purification methods to obtain artemisinin of greater than 99.9% purity. Z. Horváth, E. Horosanskaia, J. W. Lee, H. Lorenz, D. Kopetzki, K. Gilmore, P. H. Seeberger and A. Seidel-Morgenstern, manuscript in preparation.
- X. Fan, V. Sans, P. Yaseneva, D. D. Plaza, J. Williams and A. Lapkin, Org. Process Res. Dev., 2012, 16, 1039 CrossRef CAS.
- Recently, in situ generated ketones have been successfully reduced in flow using a NaBH4 as a 0.4 M solution in absolute ethanol, see: M. Fagnoni, F. Bonassi, A. Palmieri, S. Protti, D. Ravelli and R. Ballini, Adv. Synth. Catal., 2014, 356, 753 CrossRef CAS PubMed.
- (a) T. Noël, et al. , Chem. Sci., 2011, 2, 287 RSC; (b) J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618 CrossRef CAS PubMed.
- D. P. Schwartz and R. Reynolds, Microchem. J., 1975, 20, 50 CrossRef CAS.
- J. L. N. Fernandes, M. C. de Souza, E. C. S. Brenelli and J. A. Brenelli, Synthesis, 2009, 4058 CAS.
- The reduction capacity was measured using benzaldehyde as a model compound. The reduction column has been found to efficiently reduce aldehydes, ketones and in situ generated imines. Our efforts in this area are the subject of a future publication (K. Gilmore, S. Vukelić, D. T. McQuade, B. Koksch and P. H. Seeberger).
- R. W. Stringham and D. S. Teager, Org. Process Res. Dev., 2012, 16, 764 CrossRef CAS.
- C. Singh and P. Tiwari, Tetrahedron Lett., 2002, 43, 7235 CrossRef CAS.
- X. Liu and K. F. Jensen, Green Chem., 2013, 15, 1538 RSC.
- J. G. Kralj, H. R. Sahoo and K. F. Jensen, Lab Chip, 2007, 7, 256 RSC.
- M. P. Anthony, J. N. Burrows, S. Duparc, J. J. J. Moehrle and T. N. C. Wells, Malar. J., 2012, 11, 316 CrossRef PubMed.
- T. Brodmann, P. Koos, A. Metzger, P. Knochel and S. V. Ley, Org. Process Res. Dev., 2012, 16, 1102 CrossRef CAS.
- For example, see artemether, http://www.usp.org/sites/default/files/usp_pdf/EN/nonUSStandards/artemether.pdf.
- For example, see artesunate, http://apps.who.int/phint/en/p/docf/.
- J.-i. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of reaction parameters, purification, and NMR spectra are available free of charge, see DOI: 10.1039/c4cc05098c |
| This journal is © The Royal Society of Chemistry 2014 |