
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 and SO2 activation by a Cr–Cr quintuple bond†
Awal
Noor
,
Sadaf
Qayyum
,
Tobias
Bauer
,
Stefan
Schwarz
,
Birgit
Weber
and
Rhett
Kempe
*
Lehrstuhl Anorganische Chemie II, Universität Bayreuth, 95440 Bayreuth, Germany. E-mail: Kempe@uni-bayreuth.de
First published on 3rd September 2014
Abstract
A quintuply bonded dichromium complex stabilized by aminopyridinato ligands activates CO2 and SO2 by reducing the (formal) bond order of the metal–metal bonds. Oxygen abstraction is observed during CO2 activation. SO2 activation proceeds via formation of a unique dithionite complex/coordination. Furthermore, N2O activation was investigated and the formation of a tetrameric Cr–oxo complex was observed.
CO2 and SO2 are important molecules with, for instance, respect to global warming and acid rain formation, respectively. Thus, their emission has to be reduced with the search for utilization protocols of such molecules. Whereas SO2 is diversely used in the food industry and for the production of sulfuric acid, the utilization of CO2 as a C1-feedstock is still a highly attractive goal. CO2 is thermodynamically stable and chemically inert and needs to be activated prior to chemical conversions. Stoichiometric CO2 activation has been achieved (for instance) using complexes based on f-block metals,1 main group metal compounds,2 frustrated Lewis pairs,3 and most intensively using transition metal complexes. For the latter, mononuclear4 and bimetallic complexes5 have been investigated. In comparison, SO2 activation has been mainly achieved by mononuclear transition metal complexes.6
Stable molecules containing a quintuple bond have been reported for chromium and molybdenum.7,8 The bimetallic platform in such quintuply bonded complexes stores 10 electrons and is well suited for small molecule activation.9 In addition, the Cr complexes show unusual short metal–metal bonds with the shortest bonds [1.7056(12) Å] matching by length with long C–C bonds.10
We have a rather long ongoing interest in CO2 activation11 and quintuple bond reactivity9 and report here on the activation of CO2 and SO2 by a chromium–chromium quintuple bond. CO2 activation proceeds with C–O-bond cleavage resulting in the formation of a carbonyl complex and a chromium oxo species that differs structurally from the O2 activation product.9d SO2 activation leads to dithionite formation. Attempts to activate CO2 using quintuple bonds have failed so far.9g
The reaction of CO2 with quintuply bonded chromium dimer 1 (Scheme 1) leads to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage to give a CO bridged complex (2) (Scheme 1). Complex 2 shows evident reduction of CO2 to CO. Complex 2 can also be prepared by reaction of 1 with an excess of carbon monoxide in toluene. Following this reaction by NMR, it was found that the formation of 2 is nearly quantitative. Complex 2 is diamagnetic and the 1H NMR spectrum in benzene-d6 shows the resonance of the methyl protons of the isopropyl group as two doublets at 1.06 and 1.36 ppm with a coupling constant 3JHH = 6.8 Hz. The sharp singlet belonging to the methyl group of the 2,6-dimethylphenyl substituents appears at 2.11 ppm along with methyl protons of the co-crystallized toluene molecule. The resonance of the isopropyl CH-protons appears as a septet at 3.13 ppm. In 13C NMR, in addition to other signals, the peak for the carbon of the CO ligand is observed at 292 ppm. The infrared spectrum of complex 2 in the range of 4000–400 cm−1 shows no absorption bands in the region 2300–2100 cm−1. The strong absorption bands observed at 1924 and 1806 cm−1 can be attributed to the carbonyl ligands. In solution (toluene) signals at 1942 and 1857 cm−1 were recorded. The signals at higher wave numbers can be attributed to semi-bridged CO due to steric crowding, which is in accordance with the electronic structure of 2 (vide infra).
O bond cleavage to give a CO bridged complex (2) (Scheme 1). Complex 2 shows evident reduction of CO2 to CO. Complex 2 can also be prepared by reaction of 1 with an excess of carbon monoxide in toluene. Following this reaction by NMR, it was found that the formation of 2 is nearly quantitative. Complex 2 is diamagnetic and the 1H NMR spectrum in benzene-d6 shows the resonance of the methyl protons of the isopropyl group as two doublets at 1.06 and 1.36 ppm with a coupling constant 3JHH = 6.8 Hz. The sharp singlet belonging to the methyl group of the 2,6-dimethylphenyl substituents appears at 2.11 ppm along with methyl protons of the co-crystallized toluene molecule. The resonance of the isopropyl CH-protons appears as a septet at 3.13 ppm. In 13C NMR, in addition to other signals, the peak for the carbon of the CO ligand is observed at 292 ppm. The infrared spectrum of complex 2 in the range of 4000–400 cm−1 shows no absorption bands in the region 2300–2100 cm−1. The strong absorption bands observed at 1924 and 1806 cm−1 can be attributed to the carbonyl ligands. In solution (toluene) signals at 1942 and 1857 cm−1 were recorded. The signals at higher wave numbers can be attributed to semi-bridged CO due to steric crowding, which is in accordance with the electronic structure of 2 (vide infra).
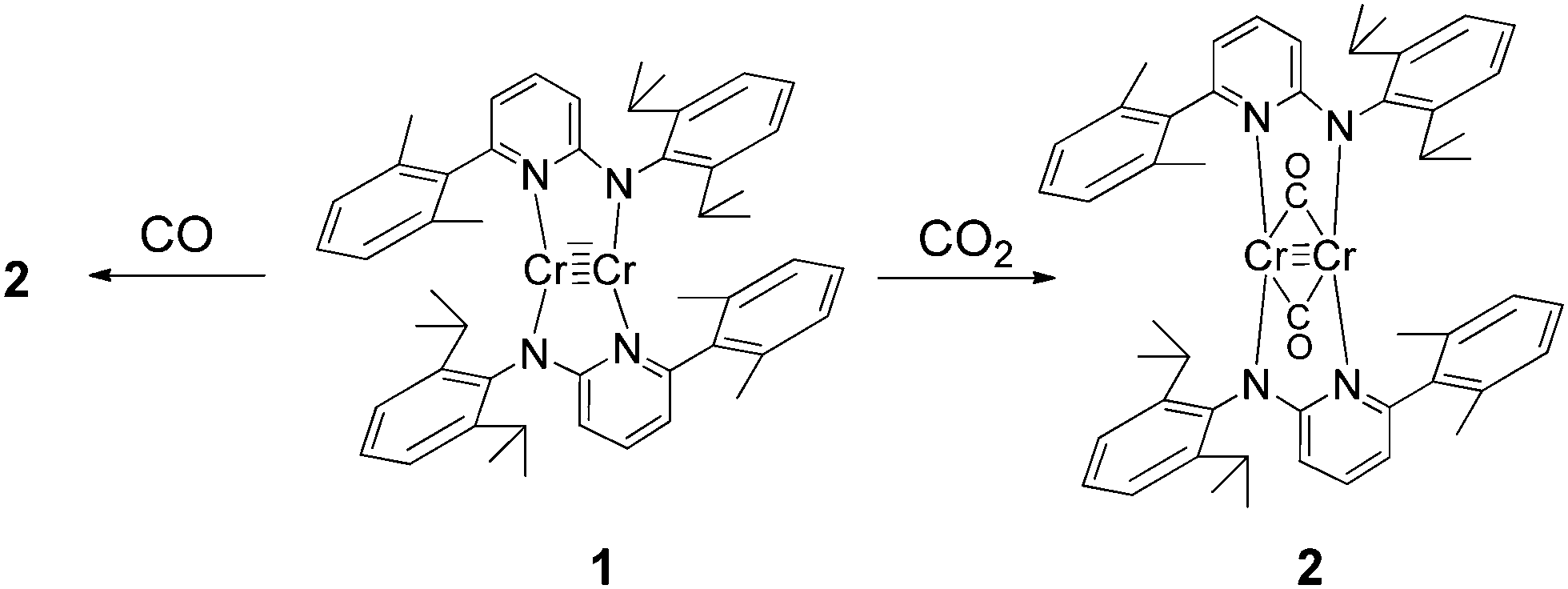 | ||
| Scheme 1 Synthesis of complex 2. In the course of the reaction with CO2, an oxo–chromium byproduct is formed (vide infra). | ||
The molecular structure of 2 is shown in Fig. 1. In 2, the two Cr atoms are in the plane of the pyridine rings of the aminopyridinato ligands. The two CO ligands are coordinated perpendicular to this plane with a C–O bond length of 1.154(8) Å and Cr–C–O angles of 152.9(6) and 153.2(6)° (Fig. 1). The Cr–C bond distances of the bridging carbonyl ligand are 2.092(8) and 2.066(8) Å. These values are comparable to Cr–Cr bonded complexes with bridging carbonyls.12 The Cr–Cr distance of 1.886(19) Å is in the range known for Cr–Cr quadruple bonds but a quadruple bond is not in accordance with the electronic structure of 2 (vide infra).
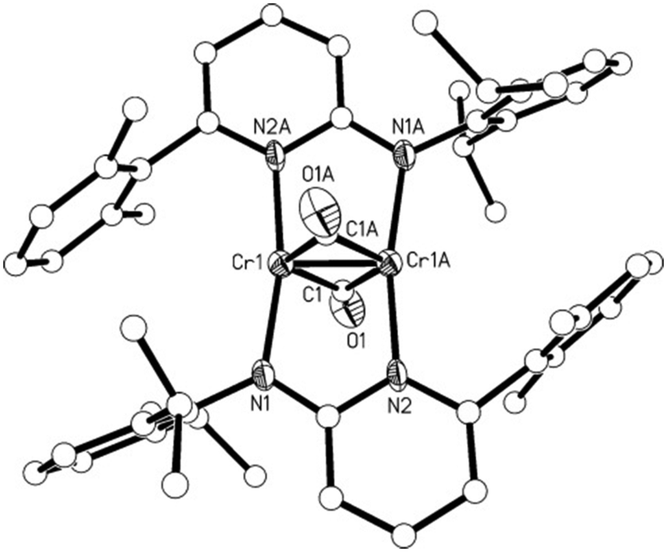 | ||
| Fig. 1 Molecular structure of 2 with hydrogen atoms and one toluene molecule omitted for clarity. Select bond lengths [Å] and angles [°]. Cr1–Cr1A 1.886(19), Cr1–N1 2.011(4), Cr1–N2 2.016(4), Cr1–C1 2.066(8), Cr1–C1 2.092(8), C1–O1 1.154(8), C1–Cr1 2.066(8); Cr1–Cr1A–N1 97.56(13), Cr1–Cr1A–N2 94.04(13), N1–Cr1–N2A 168.39(17), Cr1–Cr1A–C1 63.7(2), N1–Cr1–C1 92.0(2), N2–Cr1–C1 93.5(2), Cr1–Cr1A–C1 62.3(2), Cr1–C1–O1 152.9(6), Cr1A–C1–O1 153.2(6), C1–Cr1–C1 126.1(2), O1–C1–Cr1 152.9(6), Cr1–C1–Cr1A 53.9(2). | ||
To investigate the potential byproduct resulting from the oxygen released, NMR experiments were carried out. They showed 2 to be the major product along with some broad peaks attributable to a paramagnetic compound. This compound differs from the oxo complex isolated if 1 is reacted with O2.9d To get more insight we reacted N2O with 1 (Scheme 2). The purple solution of 1 in toluene turned green upon exposure to N2O for half an hour at room temperature. Cooling of the solution under a N2O atmosphere leads to a cluster compound 3. The X-ray structural analysis of 3 shows a chair like conformation for the central [CrO]4 fragment, in which two of the oxygens show (μ2-O) and the other two show (μ3-O) bridging coordination modes (Fig. 2). The Cr–O bond distances are as expected, longer for the (μ2-O) [1.839–2.050 Å] coordinated oxo–ligand than the (μ3-O) [1.706–1.965 Å] oxo-ligated moiety. Similarly, the Cr1–Cr1A bond length [2.7904(10) Å] for the Cr–Cr fragment joined by two (μ3-O) is shorter than the two terminal Cr–Cr fragments [2.8375(8) Å each] joined by one (μ2-O) and one (μ3-O) ligand. The aminopyridinato ligands are no more coordinated in a μ2-fashion but are coordinated in an η2-mode. The Namido–Cr bond lengths for the aminopyridinato ligands coordinated to the two central Cr atoms are comprehensively shorter [1.898(3) Å] than Namido–Cr bond lengths [2.081(1) Å] for the aminopyridinato ligands coordinated to the two terminal Cr atoms. On the other hand, Npyridine–Cr bond lengths for the two central Cr atoms are longer [2.179(3) Å] than Npyridine–Cr bond lengths [2.113(3) Å] for the two terminal Cr atoms, highlighting the flexibility of the aminopyridinato ligands.13
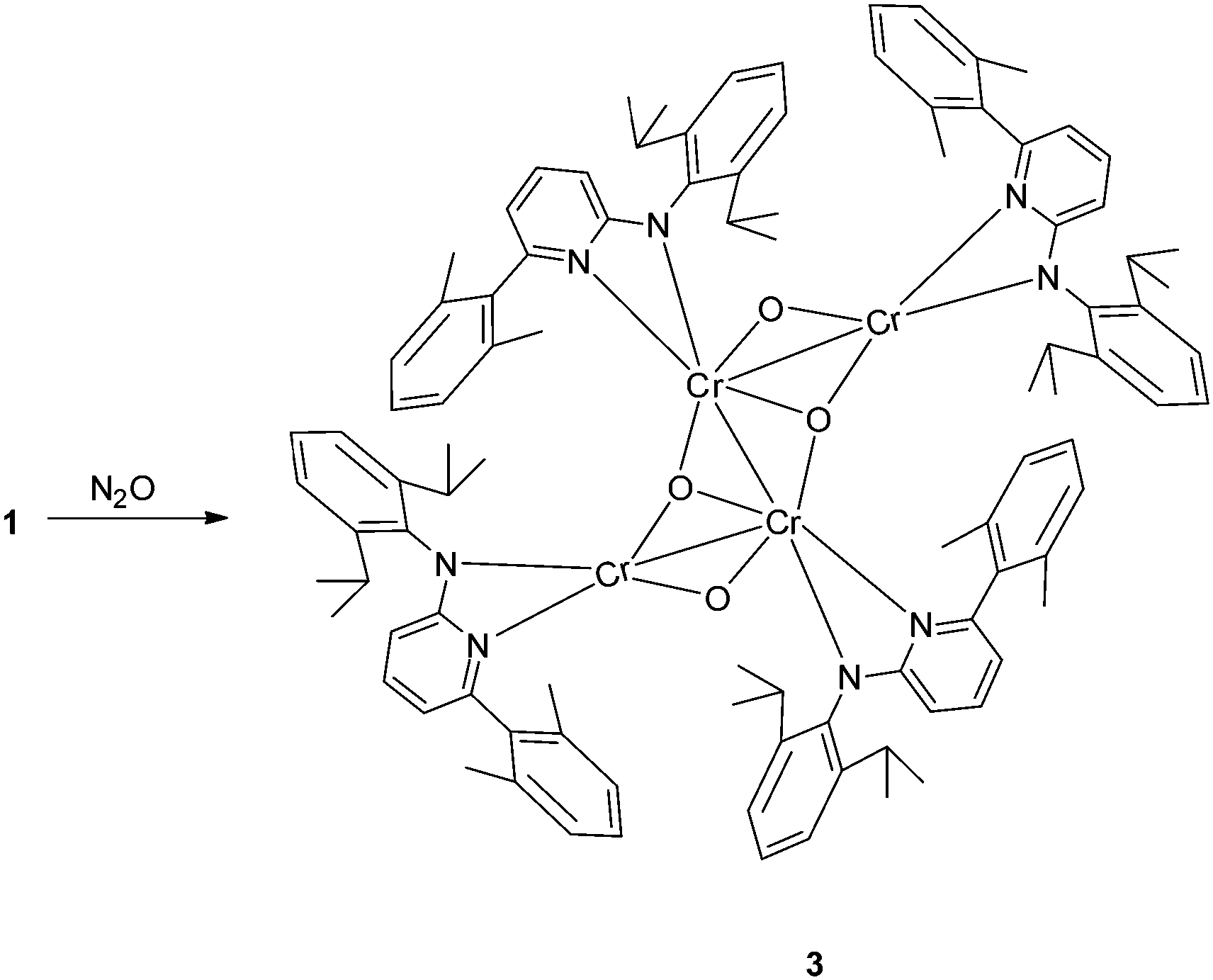 | ||
| Scheme 2 Synthesis of complex 3. | ||
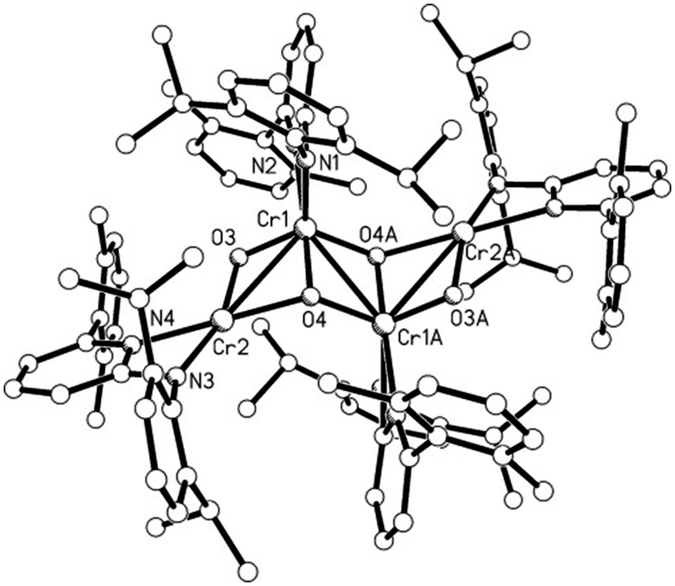 | ||
| Fig. 2 Molecular structure of 3 with hydrogen atoms omitted for clarity. Select bond lengths [Å] and angles [°]. N1–Cr1 1.898(3), N2–Cr1 2.179(3), N3–Cr2 2.081(3), N4–Cr2 2.113(3), O3–Cr1 1.702(2), O3–Cr2 1.950(2), O4–Cr1 1.840(2), O4–Cr1A 1.949(2), O4–Cr2 2.043(2), Cr1–Cr1A 2.7904(10), Cr1–Cr2 2.8375(8); Cr1–O3–Cr2 101.74(10), Cr1–O4–Cr1A 94.80(10), Cr1–O4A–Cr2A 131.82(12), Cr1–O4–Cr2 90.55(9), O3–Cr1–O4A 122.43(10), O3–Cr1–N1 119.07(11), O4–Cr1–N1 118.37(11), O3–Cr1–O4 87.55(10), O4–Cr1–O4A 85.20(10), N1–Cr1–O4A 101.29(10), O3–Cr1–N2 96.07(10), O4A–Cr1–N2 103.41(10), N1–Cr1–N2 65.75(10), O4–Cr1–N2 166.68(10), O3–Cr1–Cr1A 108.90(8), O4–Cr1–Cr1A 44.11(7), N1–Cr1–Cr1A 116.75(8), N2–Cr1–Cr1A 146.37(8), O3–Cr1–Cr2 42.29(7), O4–Cr1A–Cr2A 115.01(7), N1–Cr1–Cr2 112.13(8), N2–Cr1–Cr2 134.05(7), Cr1–Cr1A–Cr2A 78.12(2), O3–Cr2–O4 78.68(9), O3–Cr2–N3 162.07(10), O4–Cr2–N3 114.10(10), O3–Cr2–N4 102.60(10), O4–Cr2–N4 178.45(11), O3–Cr2–Cr1 35.97(6), N3–Cr2–Cr1 151.68(8), N4–Cr2–Cr1 137.74(8). | ||
The room temperature magnetic moment of 3 is 6.16, too low for a CrIII compound with four S = 3/2 centres (theoretical value: 7.74, assuming g = 2). The measured values are closer to the theoretically expected values for four S = 1 centres (μS = 5.65 μB [g = 2]) assuming some orbital momentum contribution. This is in agreement with a Cr–Cr-bond with a low bond order. Upon cooling, a slow decrease of the magnetic moment down to 3.84 μB at 25 K is observed. This behaviour is best explained with weak antiferromagnetic interactions between the remaining unpaired electrons of the four chromium centres. Indeed, the magnetic moment at 25 K is in the region expected for one unpaired electron per chromium (μS = 3.46 μB assuming g = 2). The pronounced drop in the magnetic moment below 25 K can be explained using zero field splitting.
The introduction of SO2 into a toluene solution of 1 led to an immediate colour change from purple to orange (Scheme 3). Orange crystals of 4 were grown at low temperature. X-ray analysis revealed the formation of a dithionite (S2O42−) ligand. In compound 4 the dithionite ligand lies on top of the Cr2 moiety with a bridging O2S–SO22− group where the four oxygen atoms coordinate to the two Cr atoms with Cr–O bond distances between 2.0360(14) and 2.0698(14) Å (Fig. 3). This coordination mode of dithionite to transition metals has not been described yet (to the best of our knowledge). The long S–S bond distance of 2.3786(7) Å could be due to the π antibonding interactions and is comparable to that found in Na2S2O4 [2.389 Å].14 A Cr–Cr bond distance of 2.3419(5) Å was observed. The S–O bond distances are in the range of 1.5045(15)–1.5189(15) Å. The O–S–O angles of 107.46(8) and 107.85(8)° are close to tetrahedral but the S–S–O angles (92.21(6)–93.90(6)°) deviate strongly. Compound 4 is a thiolate analogue of an oxalate, which has been previously observed due to the reductive coupling of CO2 for FeI and NiI compounds.15 The 1H NMR of 4 shows very broad peaks in the diamagnetic range of 0 to 8 ppm and could be attributed to the steric hindrance of the molecule. Low temperature experiments could not be carried out since the compound precipitates quickly in non-polar solvents like toluene and decomposes in more polar solvents like THF. Compound 4 is stable like 2 without an atmosphere of SO2 or CO2, respectively, and does not convert back to 1 as evidenced by NMR studies.
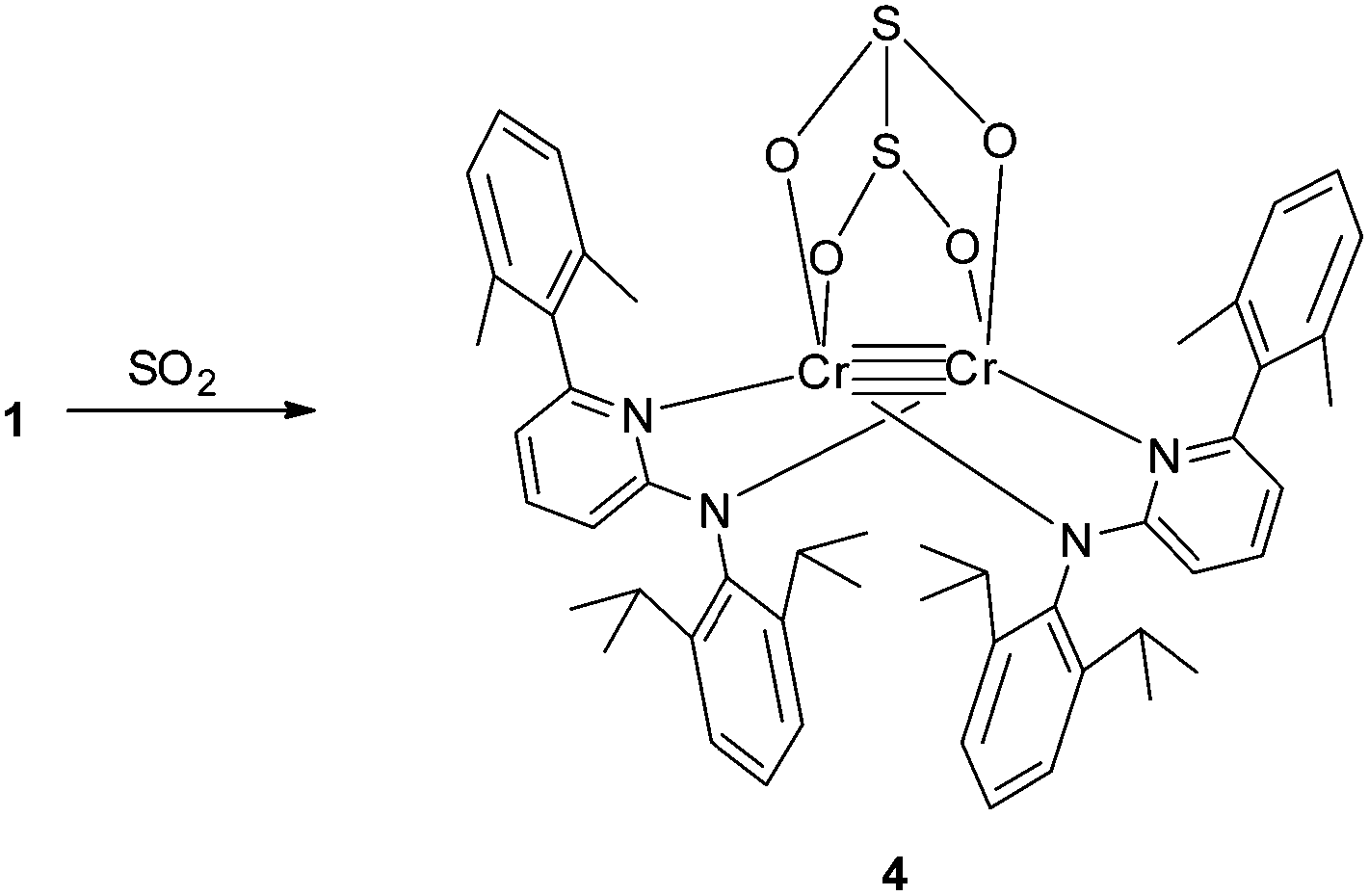 | ||
| Scheme 3 Synthesis of 4. | ||
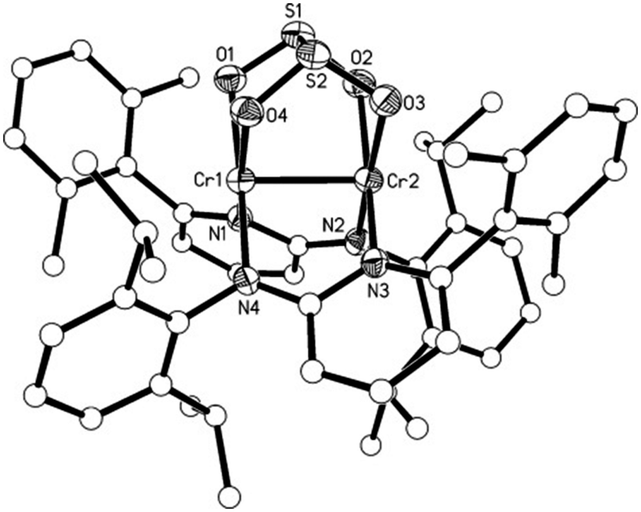 | ||
| Fig. 3 Molecular structure of 4 with hydrogen atoms omitted for clarity. Select bond lengths [Å] and angles [°]. N1–Cr1 2.0551(16), N2–Cr2 2.0594(16), O1–S1 1.5045(15), O1–Cr1 2.0360(14), O2–S1 1.5189(15), O2–Cr2 2.0698(14), S1–S2 2.3786(7), Cr1–Cr2 2.3419(5); S1–O1–Cr1 112.80(8), S1–O2–Cr2 114.73(8), S2–O3–Cr2 112.26(8), S2–O4–Cr1 115.36(8), O1–S1–O2 107.85(8), O1–S1–S2 92.21(6), O1–Cr1–N4 172.07(6), O1–Cr1–N1 89.23(6), N4–Cr1–N1 98.19(6), O1–Cr1–O4 76.65(6), N4–Cr1–O4 96.11(6), N1–Cr1–O4 165.40(6), O1–Cr1–Cr2 95.14(4), N1–Cr1–Cr2 90.47(5), O4–Cr1–Cr2 87.18(4), O3–Cr2–N2 172.16(6), O3–Cr2–N3 87.64(6), N2–Cr2–N3 99.76(6), N2–Cr2–O2 96.29(6), N3–Cr2–O2 163.55(6), O3–Cr2–Cr1 95.27(4), N2–Cr2–Cr1 87.39(5), O2–Cr2–Cr1 87.38(4). | ||
Density functional theory (DFT) calculations were performed using the TURBOMOLE16 programme package. The geometry of 1, 2 and 4 was optimized starting from structural data obtained by X-ray single crystal structure analysis. Computational details are given in the ESI.† Calculations indicate the reduction of the formal bond order from five to four for the SO2 activation product (4). Fig. S2 (ESI†) shows the metal centered HOMOs of 1 and 4. The formal bond order of the carbonyl complex 2 is lower than four. In addition to the consumption of the two δ-bonds, one of the π bonds is also significantly involved in CO bonding. The relevant frontier orbitals of 2 are shown in Fig. S3 (ESI†). The geometry optimization of 2 shows that the CO ligands form stronger bonds with one single Cr center (Table S4, ESI†). The energy of this non-centrosymmetric structure is 5 kcal mol−1 lower than the energy of the centrosymmetric structure. Due to the symmetry of the molecule, the crystal structure of 2 shows two indistinguishable Cr centers.
In summary, we could show that quintuply bonded dichromium complexes can activate CO2 and SO2 in a controlled fashion. Oxygen removal is observed during CO2 activation leading to a doubly CO bridged chromium complex. As an oxygen scavenger, an oxo-bridging tetrametallic Cr-complex could be identified. SO2 activation proceeds via S–S-bond formation leading to a unique dithionite complex.
Financial support from the Deutsche Forschungsgemeinschaft (DFG KE 756/20-2) is gratefully acknowledged. We thank Prof. S. Kümmel for helping with the calculations.
Notes and references
- (a) W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770–776 CrossRef CAS; (b) I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed; (c) I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242–11243 CrossRef CAS PubMed; (d) W. J. Evans, K. A. Miller and J. W. Ziller, Inorg. Chem., 2006, 45, 424–429 CrossRef CAS PubMed; (e) N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Chem. Commun., 2006, 4853–4855 RSC; (f) O. T. Summerscales, A. S. P. Frey, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2009, 198–200 RSC; (g) A.-C. Schmidt, A. V. Nizovtsev, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2012, 48, 8634–8636 RSC.
- T. W. Myers and L. A. Berben, Chem. Commun., 2013, 49, 4175–4177 RSC.
- (a) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed; (b) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796–1797 CrossRef PubMed; (c) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2011, 50, 8396–8399 CrossRef PubMed.
- (a) T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2006, 128, 12362–12363 CrossRef CAS PubMed; (b) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 2169–2171 CrossRef CAS PubMed; (c) J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2010, 133, 188–190 CrossRef PubMed; (d) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed; (e) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed; (f) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed; (g) S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed; (h) A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Organometallics, 2011, 30, 4308–4314 CrossRef CAS PubMed; (i) D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed; (j) C. Kleeberg, M. S. Cheung, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 19060–19063 CrossRef CAS PubMed.
- (a) A. F. R. Kilpatrick and G. F. G. Cloke, Chem. Commun., 2014, 50, 2769–2771 RSC; (b) J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21–23 CrossRef CAS; (c) T. A. Hanna, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 11363–11364 CrossRef CAS; (d) J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS PubMed; (e) L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS; (f) H. Memmler, U. Kauper, L. H. Gade, I. J. Scowen and M. McPartlin, Chem. Commun., 1996, 1751–1752 RSC; (g) T. A. Hanna, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 11363–11364 CrossRef CAS.
- (a) G. J. Kubas and R. R. Ryan, Inorg. Chem., 1984, 23, 3181–3183 CrossRef CAS; (b) G. J. Kubas, H. J. Wasserman and R. R. Ryan, Organometallics, 1985, 4, 2012–2021 CrossRef CAS; (c) K. A. Kubat-Martin, G. J. Kubas and R. R. Ryan, Organometallics, 1989, 8, 1910–1915 CrossRef CAS; (d) G. J. Kubas and R. R. Ryan, J. Am. Chem. Soc., 1985, 107, 6138–6140 CrossRef CAS; (e) G. J. Kubas, H. J. Wasserman and R. R. Ryan, Organometallics, 1985, 4, 419–421 CrossRef CAS.
- For selected reviews please see: (a) F. R. Wagner, A. Noor and R. Kempe, Nat. Chem., 2009, 1, 529–536 CrossRef CAS PubMed; (b) N. V. S. Harisomayajula, A. K. Nair and Yi.-C. Tsai, Chem. Commun., 2014, 50, 3391–3412 RSC; (c) A. K. Nair, N. V. S. Harisomayajula and Yi.-C. Tsai, Dalton Trans., 2014, 43, 5618–5638 RSC.
- (a) T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed; (b) K. A. Kreisel, G. P. A. Yap, O. Dmitrenko, C. R. Landis and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 14162–14163 CrossRef CAS PubMed; (c) R. Wolf, C. Ni, T. Nguyen, M. Brynda, G. J. Long, A. D. Sutton, R. C. Fischer, J. C. Fettinger, M. Hellman, L. Pu and P. P. Power, Inorg. Chem., 2007, 46, 11277–11290 CrossRef CAS PubMed; (d) A. Noor, F. R. Wagner and R. Kempe, Angew. Chem., 2008, 120, 7356–7359 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7246–7249 ) CrossRef PubMed; (e) Y.-C. Tsai, C.-W. Hsu, J.-S. K. Yu, G.-H. Lee, Y. Wang and T.-S. Kuo, Angew. Chem., 2008, 120, 7360–7363 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7250–7253 ) CrossRef PubMed; (f) C.-W. Hsu, J.-S. K. Yu, C.-H. Yen, G.-H. Lee, Y. Wang and Y.-C. Tsai, Angew. Chem., 2008, 120, 10081–10084 ( Angew. Chem., Int. Ed. , 2008 , 47 , 9933–9936 ) CrossRef PubMed; (g) A. Noor and R. Kempe, Chem. Rec., 2010, 10, 413–416 CAS; (h) A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko and R. Kempe, Z. Anorg. Allg. Chem., 2009, 635, 1149–1152 CrossRef CAS PubMed; (i) L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142–13148 CrossRef CAS PubMed.
- (a) Y.–L. Huang, D.-Y. Lu, H.-C. Yu, J.-S. K. Yu, C.-W. Hsu, T.-S. Kuo, G.-H. Lee, Y. Wang and Y.-C. Tsai, Angew. Chem., 2012, 124, 7901–7905 ( Angew. Chem., Int. Ed. , 2012 , 51 , 7781–7785 ) CrossRef PubMed; (b) Y.-C. Tsai, H.-Z. Chen, C.-C. Chang, J.-S. K. Yu, G.-H. Lee, Y. Wang and T.-S. Kuo, J. Am. Chem. Soc., 2009, 131, 12534–12535 CrossRef CAS PubMed; (c) S.-C. Liu, W.-L. Ke, J.-S. K. Yu, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., 2012, 124, 6500–6503 ( Angew. Chem., Int. Ed. , 2012 , 51 , 6394–6397 ) CrossRef PubMed; (d) A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko and R. Kempe, Nat. Chem., 2009, 1, 322–325 CrossRef CAS; (e) A. Noor, E. S. Tamne, S. Qayyum, T. Bauer and R. Kempe, Chem. – Eur. J., 2011, 17, 6900–6903 CrossRef CAS PubMed; (f) C. Schwarzmaier, A. Noor, G. Glatz, M. Zabel, A. Y. Timoshkin, B. M. Cossairt, C. C. Cummins, R. Kempe and M. Scheer, Angew. Chem., 2011, 123, 7421–7424 ( Angew. Chem., Int. Ed. , 2011 , 50 , 7283–7286 ) CrossRef PubMed; (g) C. Ni, B. D. Ellis, G. J. Long and P. P. Power, Chem. Commun., 2009, 2332–2334 RSC; (h) J. Shen, G. P. A. Yap, J.-P. Werner and K. H. Theopold, Chem. Commun., 2011, 47, 12191–12193 RSC; (i) H.-Z. Chen, S.-C. Liu, C.-H. Yen, J.-S. K. Yu, Y.-J. Shieh, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., 2012, 124, 10488–10492 ( Angew. Chem., Int. Ed. , 2012 , 51 , 10342–10346 ) CrossRef PubMed; (j) H.-G. Chen, H.-W. Hsueh, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., 2013, 125, 10446–10450 ( Angew. Chem., Int. Ed. , 2013 , 52 , 10256–10260 ) CrossRef PubMed; (k) M. Carrasco, N. Curado, C. Maya, R. Peloso, A. Rodríguez, E. Ruiz, S. Alvarez and E. Carmona, Angew. Chem., 2013, 125, 3309–3313 ( Angew. Chem., Int. Ed. , 2013 , 52 , 3227–3231 ) CrossRef PubMed; (l) J. Shen, G. A. Yap and K. H. Theopold, Chem. Commun., 2014, 50, 2579–2581 RSC; (m) J. Shen, G. P. A. Yap and K. H. Theopold, J. Am. Chem. Soc., 2014, 136, 3382–3384 CrossRef CAS PubMed; (n) M. Carrasco, N. Curado, E. Alvarez, C. Maya, R. Peloso, M. L. Poveda, A. Rodríguez, E. Ruiz, S. Alvarez and E. Carmona, Chem. – Eur. J., 2014, 20, 6092–6102 CrossRef CAS PubMed.
- A. Noor, T. Bauer, T. K. Todorova, B. Weber, L. Gagliardi and R. Kempe, Chem. – Eur. J., 2013, 19, 9825–9832 CrossRef CAS PubMed.
- (a) D. Walther, U. Ritter, R. Kempe, J. Sieler and B. Undeutsch, Chem. Ber., 1992, 125, 1529–1536 CrossRef CAS PubMed; (b) D. Walther, G. Bräunlich, R. Kempe and J. Sieler, J. Organomet. Chem., 1992, 436, 109–119 CrossRef CAS; (c) R. Kempe, J. Sieler, D. Walther, J. Reinhold and K. Rommel, Z. Anorg. Allg. Chem., 1993, 619, 1105–1110 CrossRef CAS PubMed.
- (a) P. Klüfers, L. Knoll, C. Reiners and K. Reiß, Chem. Ber., 1985, 118, 1825–1835 CrossRef PubMed; (b) W. Chen, L. Y. Goh and T. C. W. Mak, Organometallics, 1986, 5, 1997–2002 CrossRef; (c) L. Y. Goh and T. C. W. Mak, J. Organomet. Chem., 1989, 363, 77–89 CrossRef CAS.
- S. Deeken, G. Motz and R. Kempe, Z. Anorg. Allg. Chem., 2007, 633, 320–325 CrossRef CAS PubMed.
- J. D. Dunitz, Acta Crystallogr., 1956, 9, 579–586 CrossRef CAS.
- (a) C. C. Lu, C. T. Saouma, M. W. Day and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 4–5 CrossRef CAS PubMed; (b) C. T. Saouma, C. C. Lu, M. Day and J. C. Peters, Chem. Sci., 2013, 4, 4042–4051 RSC; (c) B. Horn, C. Limberg, C. Herwig and B. Braun, Chem. Commun., 2013, 49, 10923–10925 RSC.
- TURBOMOLE V6.4 2012, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
Footnote |
| † Electronic supplementary information (ESI) available: General and synthetic methods, instrumentation, and full crystallographic data. CCDC 1009949 (2), 1009951 (3) and 1009950 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc05071a |
| This journal is © The Royal Society of Chemistry 2014 |