
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative skeletal rearrangement of 1,1′-binaphthalene-2,2′-diamines (BINAMs) via C–C bond cleavage and nitrogen migration: a versatile synthesis of U-shaped azaacenes†
Youhei
Takeda
*ab,
Masato
Okazaki
b and
Satoshi
Minakata
*b
aFrontier Research Base for Global Young Researchers, Graduate School of Engineering, Osaka University, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan. E-mail: takeda@chem.eng.osaka-u.ac.jp; minakata@chem.eng.osaka-u.ac.jp
First published on 17th July 2014
Abstract
An oxidative skeletal rearrangement of 1,1′-binaphthalene-2,2′-diamines (BINAMs) that involves the cleavage of a strong C–C single bond of the binaphthalene unit and the nitrogen migration has been discovered. The unprecedented rearrangement enables access to a series of U-shaped azaacenes otherwise difficult to prepare in a selective manner by classical methods. Moreover, physicochemical properties of the unique azaacenes have been comprehensively investigated.
Azaacenes, in which parts of C
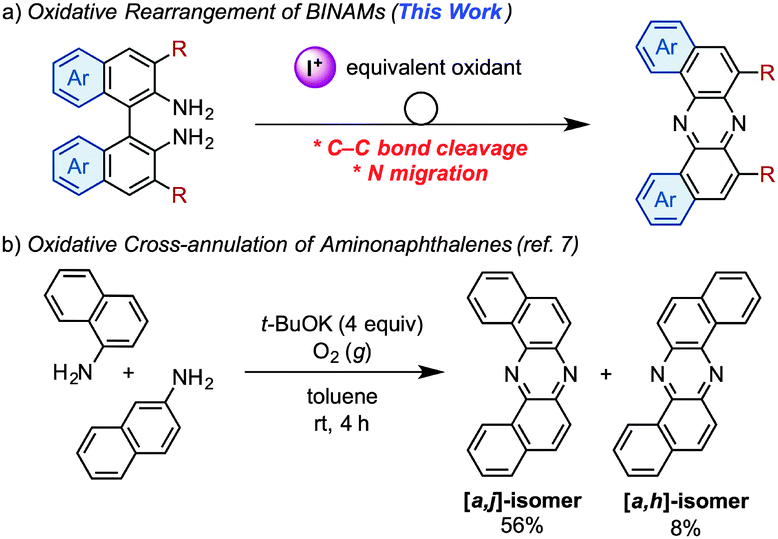
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH fragments of acenes are formally replaced with isoelectronic imine (CN) moieties, have been emerging as promising candidates for electron-transporting (n-type) materials in organic electronics.1 The introduction of electronegative nitrogen atoms into π-conjugated carbon frameworks allows increasing electron affinities (EAs) to lower the barriers for electron injection from metal electrodes.2 Furthermore, owing to the decrease in the number of C–H⋯π contacts, azaacenes show a high propensity for adopting densely stacked packing-structures in the solid states, which favor a carrier-transporting process.3 However, due to limited synthetic accessibility of azaacenes, the exploration of azaacene-based materials has fatally lagged behind those of acenes. Therefore, the development of novel synthetic approaches to azaacenes is fundamentally important from the viewpoints of not only synthetic chemistry but also materials science. Traditionally, syntheses of azaacenes have exclusively relied on condensation/oxidation protocols starting from ortho-arylenediamines and ortho-quinone derivatives.4 In this context, recently, a new synthetic protocol toward the preparation of azaacenes consisting of a Pd-catalyzed double amination of activated ortho-dichloroarenes with ortho-arylenediamines and a successive oxidation leading to linear azaacenes in an efficient manner, has been reported.5 On the other hand, Hiroto and Shinokubo have reported a DDQ-promoted oxidative annulation of aromatic amines to selectively give kinked azaacenes and aza[7]helicenes depending on the conditions applied.6 Herein we present a new strategy to synthesize azaacenes based on the discovery of an unprecedented oxidative skeletal rearrangement of 1,1′-binaphthalene-2,2′-diamines (BINAMs) induced by an iodine-containing oxidant, which exclusively provides a series of dibenzo[a,j]phenazine-cored U-shaped azaacenes (Scheme 1a). As a relevant work, there is a single report that describes selective preparation of dibenzo[a,j]phenazine through an oxidative cross-annulation of α- and β-aminonaphthalenes mediated by a strong base (t-BuOK) under an O2 atmosphere (Scheme 1b).7 However, this known method intrinsically requires two regioisomers of aminonaphthalenes and suffers from the concomitant production of a constitutional [a,h]-isomer, thereby lacking the versatility as a synthetic approach to U-shaped azaacenes.
CH fragments of acenes are formally replaced with isoelectronic imine (CN) moieties, have been emerging as promising candidates for electron-transporting (n-type) materials in organic electronics.1 The introduction of electronegative nitrogen atoms into π-conjugated carbon frameworks allows increasing electron affinities (EAs) to lower the barriers for electron injection from metal electrodes.2 Furthermore, owing to the decrease in the number of C–H⋯π contacts, azaacenes show a high propensity for adopting densely stacked packing-structures in the solid states, which favor a carrier-transporting process.3 However, due to limited synthetic accessibility of azaacenes, the exploration of azaacene-based materials has fatally lagged behind those of acenes. Therefore, the development of novel synthetic approaches to azaacenes is fundamentally important from the viewpoints of not only synthetic chemistry but also materials science. Traditionally, syntheses of azaacenes have exclusively relied on condensation/oxidation protocols starting from ortho-arylenediamines and ortho-quinone derivatives.4 In this context, recently, a new synthetic protocol toward the preparation of azaacenes consisting of a Pd-catalyzed double amination of activated ortho-dichloroarenes with ortho-arylenediamines and a successive oxidation leading to linear azaacenes in an efficient manner, has been reported.5 On the other hand, Hiroto and Shinokubo have reported a DDQ-promoted oxidative annulation of aromatic amines to selectively give kinked azaacenes and aza[7]helicenes depending on the conditions applied.6 Herein we present a new strategy to synthesize azaacenes based on the discovery of an unprecedented oxidative skeletal rearrangement of 1,1′-binaphthalene-2,2′-diamines (BINAMs) induced by an iodine-containing oxidant, which exclusively provides a series of dibenzo[a,j]phenazine-cored U-shaped azaacenes (Scheme 1a). As a relevant work, there is a single report that describes selective preparation of dibenzo[a,j]phenazine through an oxidative cross-annulation of α- and β-aminonaphthalenes mediated by a strong base (t-BuOK) under an O2 atmosphere (Scheme 1b).7 However, this known method intrinsically requires two regioisomers of aminonaphthalenes and suffers from the concomitant production of a constitutional [a,h]-isomer, thereby lacking the versatility as a synthetic approach to U-shaped azaacenes.
 | ||
| Scheme 1 Construction of dibenzophenazine-based U-shaped azaacenes. | ||
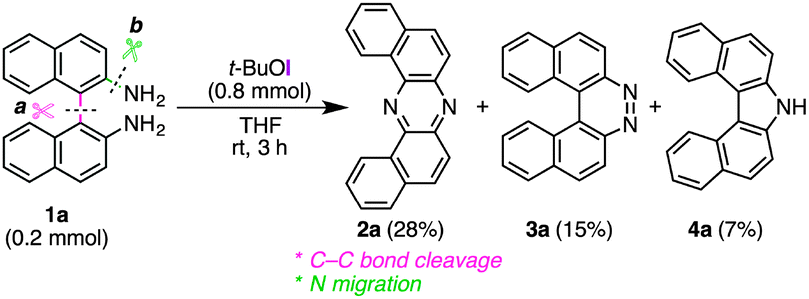
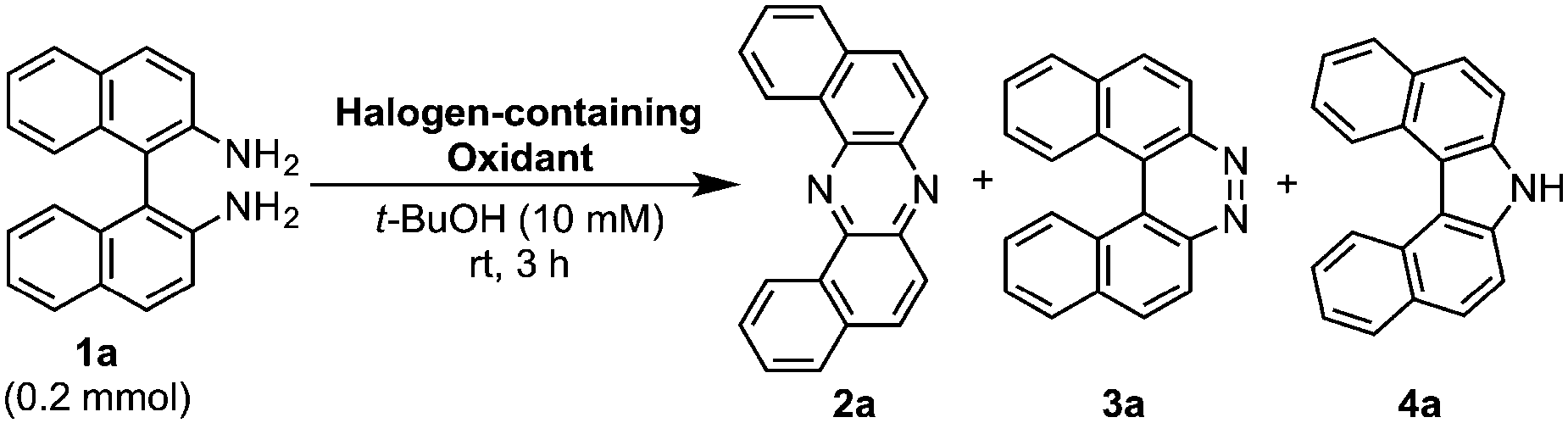
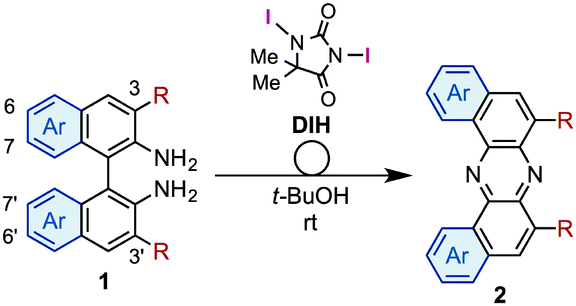
During our studies on the development of oxidative transformations of various amines utilizing iodine-containing oxidants,8 we serendipitously found out that the treatment of BINAM (1a) with tert-butyl hypoiodite (t-BuOI)9 produced dibenzo[a,j]phenazine (2a) in 28% yield along with the production of reasonably expected compounds 3a10 (15%) and 4a11 (7%) [eqn (1)].
 | (1) |
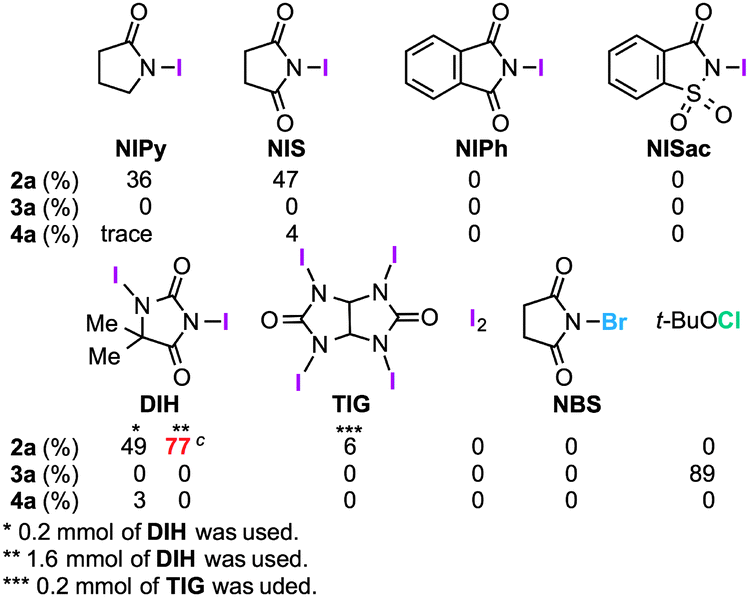
The structure of 2a was unambiguously confirmed by the X-ray crystallographic analysis of its yellow single crystal (ESI†). Most importantly, the discovered transformation leading to 2a formally involves the cleavage of the strong C(Ar)–C(Ar) bond [a in eqn (1)] of the binaphthyl unit (e.g., the bond dissociation energy (BDE) of Ph–Ph is as large as 118 kcal mol−1)12 without the aid of transition metal complexes13 and also involves the migration of a nitrogen atom [b in eqn (1)] to the adjacent carbon. Encouraged by this finding, we envisioned that the utilization of this unique rearrangement reaction would offer a versatile route to U-shaped azaacenes. To establish a novel approach to synthesize U-shaped azaacenes, reaction parameters were scrutinized to identify the conditions to selectively provide 2a (ESI†). Table 1 summarizes the effect of halogen-containing oxidants. To our delight, the use of N-iodolactams as oxidants like NIS, NIPy, and DIH in t-BuOH selectively gave 2a in moderate yields along with trace amounts of 4a without affording 3a. The use of 8 mol equivalents of DIH gave 2a in 77% yield, although the role of excess of DIH is not clear at present. In sharp contrast, when N-chloro-containing oxidant (t-BuOCl) was used, diaza[5]helicene 3a was exclusively formed in a high yield (89%). Other types of oxidants like I2, NBS, DDQ, PhI(OAc)2, and MnO2 failed to furnish 2a.
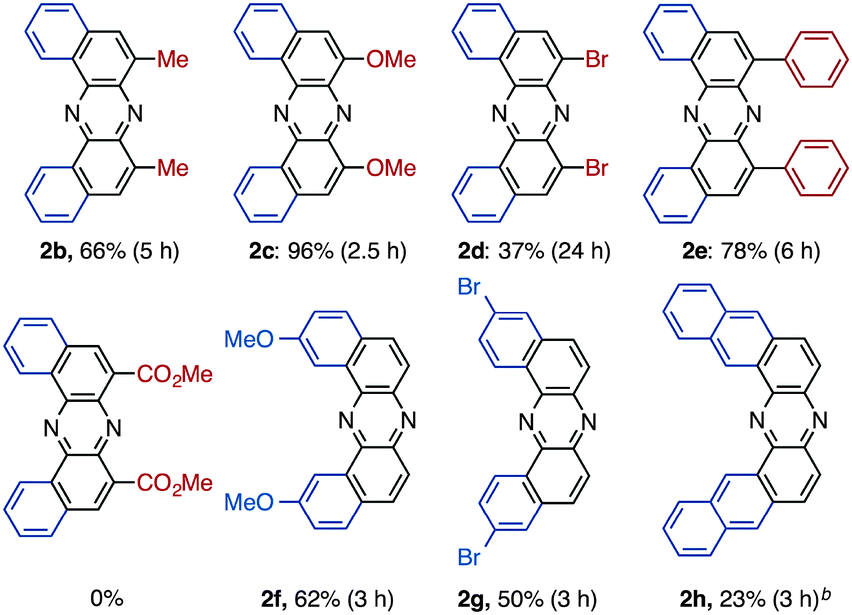
Having identified the optimal conditions for the rearrangement reaction, the scope and limitation of BINAM substrates were surveyed (Table 2). The treatment of BINAMs bearing two electron-donating substituents (Me and MeO) at the 3,3′-position with DIH successfully provided azaacenes 2b and 2c in 66% and 96% yields, respectively. The reaction using 3,3′-dibrominated BINAM 1d gave desired product 2d in a low yield, probably because of the poor solubility of the diamine in t-BuOH. Notably, the rearrangement of 3,3′-diphenyl diamine 1e also efficiently proceeded to afford 2e in a high yield, indicating that steric hindrance around the amino moieties does not affect the reaction efficiency. In sharp contrast, diamine bearing two ester groups (CO2Me) at the 3,3′-position did not undergo the rearrangement at all. This significant lack of reactivity might be ascribed to the low nucleophilicity of amino moieties, inhibiting the formation of N–I bonds (vide infra). Diamines bearing the 7,7′- and 6,6′-disubstituent also underwent rearrangement to selectively provide 2f and 2g in moderate yields. Notably, the rearrangement of bianthracene diamine 1h gave highly conjugated heptacyclic azaacene 2h, which should be quite difficult to synthesize by conventional organic reactions.
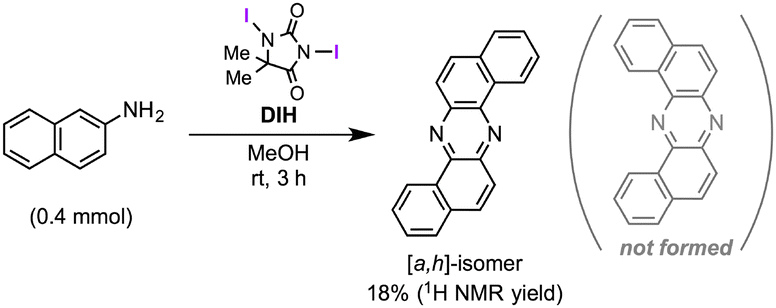
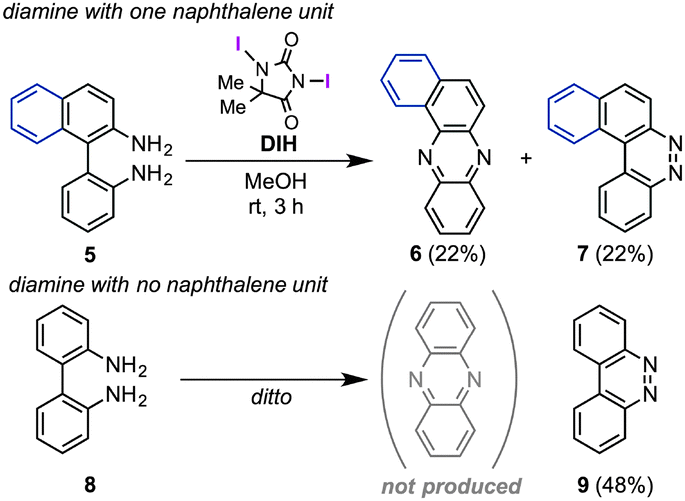
Regarding mechanistic aspects, importantly, the existence of at least one naphthalene unit is indispensable for the unique rearrangement (Scheme 2): when biaryldiamines bearing one (5) or no (8) naphthyl unit were subjected to the reaction conditions, the ratios of rearranged products/benzocinnoline products were found to be positively correlated with the number (n) of naphthalene units. These results might indicate the involvement of dearomatization processes in the reaction pathway (vide infra), which would be more favorable with naphthalene units than benzene units.14 On the other hand, a cross-over experiment applying an equimolar mixture of 1a and 1c resulted in no production of cross-products at all (Scheme 3), and a control experiment using β-naphthylamine under the optimal conditions led to the production of the [a,h]-isomer without the formation of 2a [eqn (2)], suggesting the intramolecularity of the rearrangement.
 | (2) |
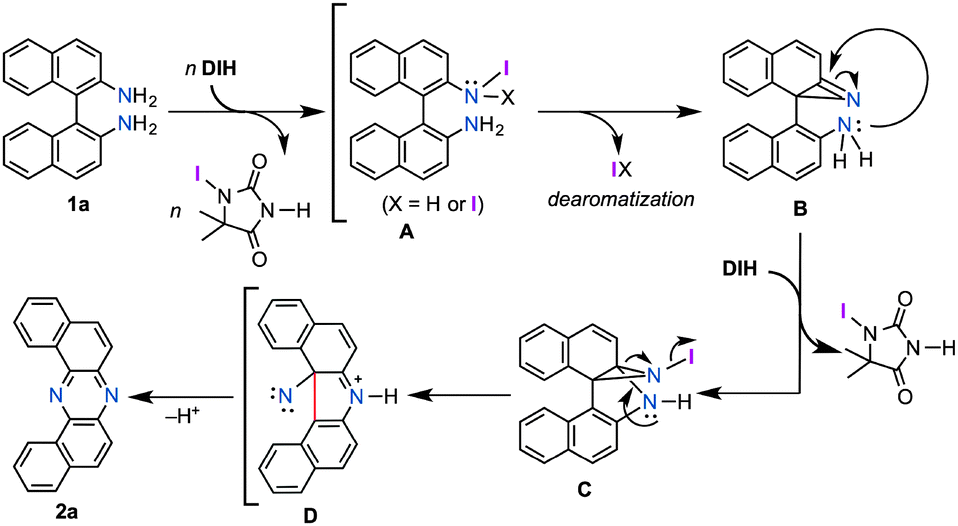
Although a precise mechanism should await further investigation, in conjunction with the accumulated knowledge of the reactivities of aromatic amines toward electrophilic iodinating oxidants, a tentative reaction pathway is illustrated in Fig. 1. The reaction would start with exchange of N–H hydrogen(s) with iodine, leading to mono or di N-iodoamine A.8 The dearomatization process to form azirine B could explain the indispensability of the naphthyl unit for the rearrangement.15 Upon the formation of B, the highly strained intermediate would be attacked by an intramolecular amino nucleophile (Ar–NH2), and the developing anion would be trapped by the extra iodinating reagent to form C.16 Nitrene D could be generated from C driven by the release of its strain energy, and the subsequent insertion of nitrene to the C(Ar)–C(Ar) bond would give phenazine 2a. Alternatively, the path from C to 2a could be interpreted as a variant of Stieglitz rearrangement.17 Carbazole 4a might be produced through a [3,3]-sigmatropic rearrangement of the diimine that would be formed by the protonation of 1a with generated HI, and followed by the elimination of ammonia.11
 | ||
| Fig. 1 A tentative pathway of the rearrangement. | ||
 | ||
| Scheme 2 Influence of a naphthalene unit. | ||
 | ||
| Scheme 3 Cross-over experiment. | ||
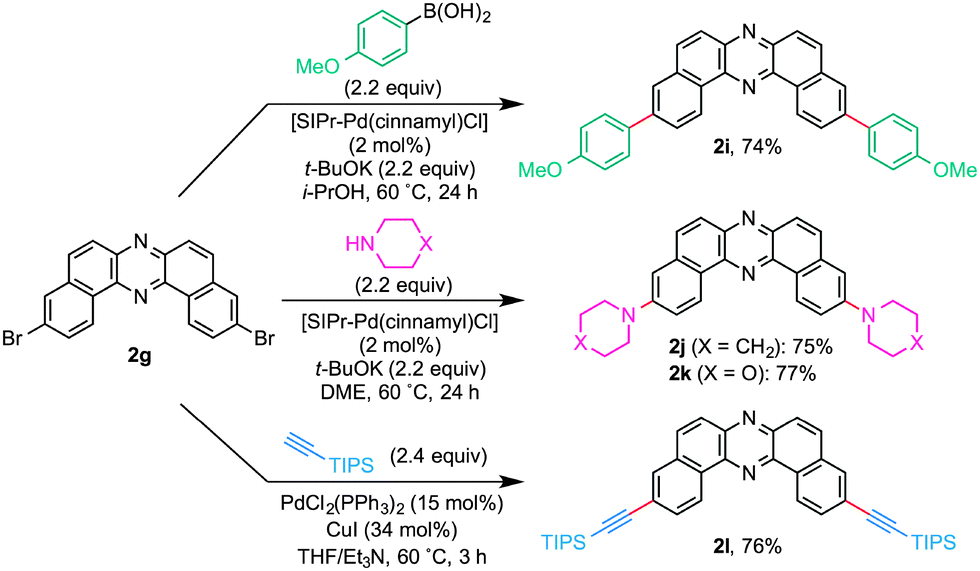
In light of bromo functionality of 2d and 2g, these compounds should serve as useful building blocks for functionalized azaacenes. Synthetic versatility of 2g was clearly demonstrated by the preparation of 2i–2lvia Pd catalysis (Scheme 4). With slight modifications of the Nolan’s original conditions,18 NHC/Pd-catalyzed cross-coupling of 2g with an arylboronic acid and cyclic amines gave the corresponding coupled products 2i–2k in good yields. Furthermore, TIPS-acetylene was efficiently cross-coupled with 2g to afford conjugation-extended azaacene 2l in 76% yield.
 | ||
| Scheme 4 Pd-catalyzed functionalization of 2g. | ||
Basic physicochemical properties of U-shaped azaacenes 2 were investigated. UV/Vis and fluorescence (FL) spectra as well as a cyclic voltammogram for 2a are shown in Fig. 2 as a representative example (for other compounds, see the ESI†). The mirror-image type UV and FL spectra with fine vibrational structures and a small Stokes shift (9 nm) reflects the rigid structure of 2a like typical (aza-)acenes (Fig. 2a). Likewise, diluted CH2Cl2 solutions of other azaacenes 2 emit fluorescence ranging from blue (λem = 425 nm) to yellow (λem = 561 nm) depending on the substituents on the conjugated core. Especially, the introduction of sterically demanding and strongly electron-donating groups like piperidino and morpholino functionalities (i.e., 2j and 2k) resulted in significant Stokes shifts and greatly enhanced quantum yields (2a: λem = 425 nm, Φf 0.14; 2j: λem = 561 nm, Φf 0.47; 2k: λem = 543 nm, Φf 0.42), probably ascribed to intramolecular charge-transfer (ICT) emission. Notably, most of the azaacenes 2 showed one pair of reversible redox waves at the potentials ranging from −1.76 to −1.98 V against the Fc/Fc+ redox couple, as shown in Fig. 2b, indicating the good electron-accepting abilities of 2. These values are compatible with those of n-type organic materials used for organic luminescence diode (OLED) devices like diarylanthrazolines19 and Alq3.20
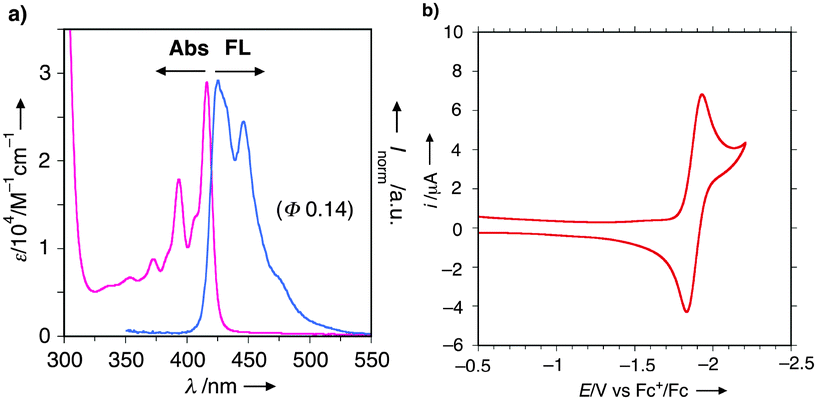 | ||
| Fig. 2 (a) UV/Vis absorption and FL spectra of CH2Cl2 solution of 2a (10−5 M order). (b) Cyclic voltammogram of CH2Cl2 solution of 2a (10−4 M order) using nBu4NPF6 as an electrolyte. | ||
In conclusion, we have discovered an unprecedented oxidative skeletal rearrangement of BINAMs and established an approach to constructing novel types of azaacenes that are otherwise difficult to synthesize by conventional methodologies. Investigation into the creation of functional conjugated materials based on these unique azaacenes are ongoing in our laboratory.
We deeply thank Professor Toshiyuki Moriuchi of Osaka University for his assistance in single-crystal X-ray diffraction experiments. This research was partly supported by the research Grants from the Izumi Science and Technology foundation and from the Murata Science Foundation, the Sasakawa Scientific Research Grant from the Japan Science Society, and the Research Encouragement Grants from The Asahi Glass Foundation (to Y.T.).
Notes and references
- For reviews on azaacenes, see: (a) U. H. F. Bunz, Chem. – Eur. J., 2009, 15, 6780 CrossRef CAS PubMed; (b) G. J. Richards, J. P. Hill, T. Mori and K. Ariga, Org. Biomol. Chem., 2011, 9, 5005 RSC; (c) Q. Miao, Synlett, 2012, 326 CrossRef CAS PubMed; (d) U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810 CrossRef CAS PubMed.
- M. Winkler and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 1805 CrossRef CAS PubMed.
- K. E. Maly, Cryst. Growth Des., 2011, 11, 5628 CAS.
- (a) S. More, R. Bhosale, S. Choudhary and A. Mateo-Alonso, Org. Lett., 2012, 14, 4170 CrossRef CAS PubMed; (b) A. Mateo-Alonso, N. Kulisic, G. Valenti, M. Marcaccio, F. Paolucci and M. Prato, Chem. – Asian J., 2010, 5, 482 CrossRef CAS PubMed; (c) A. L. Appleton, S. M. Brombosz, S. Barlow, J. S. Sears, J.-L. Bredas, S. R. Marder and U. H. F. Bunz, Nat. Commun., 2010, 1, 91 CrossRef PubMed; (d) B. Gao, M. Wang, Y. Cheng, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2008, 130, 8297 CrossRef CAS PubMed; (e) Y. Fogel, M. Kastler, Z. Wang, D. Andrienko, G. J. Bodwell and K. Müllen, J. Am. Chem. Soc., 2007, 129, 11743 CrossRef CAS PubMed.
- (a) O. Tverskoy, F. Rominger, A. Peters, H.-J. Himmel and U. H. F. Bunz, Angew. Chem., Int. Ed., 2011, 50, 3557 CrossRef CAS PubMed; (b) B. D. Lindner, J. U. Engelhart, O. Tverskoy, A. L. Appleton, F. Rominger, A. Peters, H.-J. Himmel and U. H. F. Bunz, Angew. Chem., Int. Ed., 2011, 50, 8588 CrossRef CAS PubMed.
- (a) M. Akita, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 2894 CrossRef CAS PubMed; (b) K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 10333 CrossRef CAS PubMed; (c) H. Yokoi, N. Wachi, S. Hiroto and H. Shinokubo, Chem. Commun., 2013, 50, 2715 RSC.
- Y. Kosugi, K. Itoho, H. Okazaki and T. Yanai, J. Org. Chem., 1995, 60, 5690 CrossRef CAS.
- (a) S. Minakata, Acc. Chem. Res., 2009, 42, 1172 CrossRef CAS PubMed; (b) Y. Takeda, S. Okumura and S. Minakata, Angew. Chem., Int. Ed., 2012, 51, 7804 CrossRef CAS PubMed; (c) Y. Takeda, S. Okumura and S. Minakata, Synthesis, 2013, 1029 CAS; (d) S. Okumura, Y. Takeda, K. Kiyokawa and S. Minakata, Chem. Commun., 2013, 49, 9266 RSC; (e) S. Okumura, C.-H. Lin, Y. Takeda and S. Minakata, J. Org. Chem., 2013, 78, 12090 CrossRef CAS PubMed.
- D. D. Tanner, G. C. Gidley, N. Das, J. E. Rowe and A. Potter, J. Am. Chem. Soc., 1984, 106, 5261 CrossRef CAS.
- Production of benzo[c]cinnoline derivatives through dehydrogenation of biaryldiamines have been reported, see: (a) J. F. Corbett and P. F. Holt, J. Chem. Soc., 1961, 3695 RSC; (b) I. Bhatnagar and M. V. George, J. Org. Chem., 1968, 33, 2407 CrossRef CAS; (c) L. Racané, H. Čičak, Z. Mihalić, G. Karminski-Zamola and V. Tralić-Kulenović, Tetrahedron, 2011, 67, 2760 CrossRef PubMed.
- Production of carbazoles from biaryldiamines under oxidative or acidic conditions have been reported, see: (a) J. Cornforth, D. D. Ridley, A. F. Sierakowski, D. Uguen and T. W. Wallace, J. Chem. Soc., Perkin Trans. 1, 1982, 2317 RSC; (b) T. Yamato, C. Hideshima, K. Suehiro, M. Tashiro, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1991, 56, 6248 CrossRef CAS; (c) B.-Y. Lim, M.-K. Choi and C.-G. Cho, Tetrahedron Lett., 2011, 52, 6015 CrossRef CAS PubMed.
- A. A. Zavitsas, J. Phys. Chem. A, 2003, 107, 897 CrossRef CAS.
- For reviews on C–C bond activation by transition metal complexes, see: (a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (b) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (c) S. M. Bonesi and M. Fagnoni, Chem. – Eur. J., 2010, 16, 13572 CrossRef CAS PubMed.
- The loss of aromatic resonance energies for naphthalene is 11 kcal/mol smaller than that for benzene: M. B. Smith and J. March, March’s Advanced Organic Chemistry: Reactions, Mechanism, and Structure, Wiley-Interscience, New York, 5th edn, 2001 Search PubMed.
- Naphthoazirines are transiently formed from the naphthylnitrenes through the photoirradiation and thermal decomposition of naphthylazides: (a) A. Maltsev, T. Bally, M.-L. Tsao, M. S. Platz, A. Kuhn, M. Vosswinkel and C. Wentrup, J. Am. Chem. Soc., 2003, 126, 237 CrossRef PubMed; (b) N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844 CrossRef CAS PubMed . Nevertheless, neither photoirradiation and thermal decomposition of 2′-azido-[1,1′-binaphthalen]-2-amine (10) nor 2,2′-diazido-1,1′-binaphthalene (11) gave 2a, indicating that the generation of free-nitrene species in the formation of B would not likely be involved. For the detailed experimental information, see the ESI†.
- Naphthoazirines are trapped by various nucleophiles like alcohols and amines, see: (a) J. Rigaudy, C. Igier and J. Barcelo, Tetrahedron Lett., 1975, 16, 3845 CrossRef; (b) S. E. Carroll, B. Nay, E. F. V. Scriven and H. Suschitzky, Tetrahedron Lett., 1977, 18, 943 CrossRef; (c) S. E. Carroll, B. Nay, E. F. V. Scriven, H. Suschizky and D. R. Thomas, Tetrahedron Lett., 1977, 36, 3175 CrossRef; (d) B. Nay, E. F. V. Scriven, H. Suschitzky and Z. U. Khan, Synthesis, 1977, 757 CrossRef CAS.
- (a) J. Stieglitz and P. N. Leech, Chem. Ber., 1913, 46, 2147 CrossRef CAS; (b) J. Stieglitz and P. N. Leech, J. Am. Chem. Soc., 1914, 36, 272 CrossRef.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed.
- C. J. Tonzola, M. M. Alam, W. Kaminsky and S. A. Jenekhe, J. Am. Chem. Soc., 2003, 125, 13548 CrossRef CAS PubMed.
- P. E. Burrows, Z. Shen, V. Bulovic, D. M. McCarty, S. R. Forrest, J. A. Cronin and M. E. Thompson, J. Appl. Phys., 1996, 79, 7991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, NMR spectra, and physicochemical properties. CCDC 1004407. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc04911j |
| This journal is © The Royal Society of Chemistry 2014 |