
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild hypervalent iodine mediated oxidative nitration of N-aryl sulfonamides†
Ulrich
Kloeckner
and
Boris J.
Nachtsheim
*
Institute of Organic Chemistry, Eberhard Karls Universität Tübingen, 72076 Tübingen, Germany. E-mail: boris.nachtsheim@uni-tuebingen.de; Fax: +49 7071 29 5897
First published on 23rd July 2014
Abstract
An oxidative and acid-free method for the nitration of N-aryl sulfonamides has been developed using a combination of sodium nitrite as cheap and easy to handle NO2-source and the hypervalent iodine reagent PIFA as stoichiometric oxidant. Under very mild reaction conditions, the desired mononitrated aryl sulfonamides were isolated in up to 87% yield. This is the first example of an iodane-mediated oxidative nitration.
Nitroarenes are versatile structural motifs since they have been widely utilized as important precursors for the synthesis of pharmaceuticals and biological active compounds. Furthermore, nitrated arenes and heteroarenes can be found in a variety of drugs, pesticides and explosives.1 The most common approach to synthesize nitroarenes is the direct electrophilic nitration of arenes. Traditionally, this transformation requires harsh reaction conditions such as concentrated nitric acid or mixtures of nitric acid and sulphuric acid in order to generate the reactive nitronium cation (NO2+) through dehydration of nitric acid.2 As an alternative, stoichiometric amounts of metal nitrates such as Bi(NO)3 or expensive nitronium salts can be applied.3 Even though these methods turned out to be mild alternatives to classical nitration conditions, they are usually restricted to reactive, electron rich arenes and unfortunately, stoichiometric amounts of metal oxides are generated as undesired side products. Recently, tert-butyl nitrite (TBN) was found by Savinov and co-workers to be a mild nitration reagent for phenols.4–6 Subsequently, this reagent was used extensively for oxidative radical nitrations of alkenes and nitration cascade reactions.7–13 In 2013 Arns reported an elegant chemoselective nitration of N-aryl sulfonamides using tert-butyl nitrite.14 Despite its great versatility, TBN is potential hazardous due to its known highly exothermic decomposition (1200 J g−1) at elevated temperatures (above 110 °C).15
A so far neglected way to generate nitronium cations under non-acidic conditions would be a direct 1- or 2-electron oxidation of sodium nitrite.16 In this communication we want to describe such a reaction mediated by aryl-λ3-iodanes as mild and safe oxidants and atom transfer reagents.17–25 Aryl-λ3-iodanes of type ArIX2 readily undergo ligand exchange reactions to substitute one or both ligands X against external anions or neutral nucleophiles (Scheme 1). We proposed that ligand exchange with sodium nitrite should give the highly reactive key intermediate A which should subsequently decompose in a 2-electron transfer process to generate phenyl iodide and a nitronium cation (Scheme 1a). As an alternative, a radical mechanism can be proposed through generation of NO2 (Scheme 1b). However, herein we want to report the development of such an oxidative transformation, which is not only a remarkable mild alternative to classical nitration reactions, but is also, to the best of our knowledge, the first example of a hypervalent iodine mediated oxidative nitration.
 | ||
| Scheme 1 Proposed oxidative generation of nitronium ions (a) or NO2 (b) using aryl-λ3-iodanes. | ||
To start our investigations, we chose to use N-phenyl sulfonamide 1a as a comparatively unreactive arene. We were pleased to find that our first attempt using a combination of the aryl-λ3-iodane PIFA (phenyl iodine bis(trifluoroacetate)) and NaNO2 in DMSO at room temperature already resulted in formation of the desired nitrated N-phenyl sulfonamide 2a in 55% yield with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 mixture of the ortho and para isomers (Table 1, entry 1). Addition of more than 1.2 equivalents of either PIFA or NaNO2 did not result in improved yields or regioselectivities (data not shown). Therefore, we concentrated our further optimization on solvent and temperature. In ethyl acetate and DMF the yields could be further improved to 85% (Table 1, entries 2 and 3). Finally, acetonitrile turned out to be the solvent of choice giving 2a in 87% yield. However, it has to be mentioned that o/p selectivity dropped slightly (Table 1, entry 4).
:1.4 mixture of the ortho and para isomers (Table 1, entry 1). Addition of more than 1.2 equivalents of either PIFA or NaNO2 did not result in improved yields or regioselectivities (data not shown). Therefore, we concentrated our further optimization on solvent and temperature. In ethyl acetate and DMF the yields could be further improved to 85% (Table 1, entries 2 and 3). Finally, acetonitrile turned out to be the solvent of choice giving 2a in 87% yield. However, it has to be mentioned that o/p selectivity dropped slightly (Table 1, entry 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | [O] | Time (h) | Temp. (°C) | Ratio (o:p) |
Yield (%) |
| a Reaction conditions: 0.2 mmol 1a, 0.24 mmol NaNO2, 0.24 mmol PIFA or PIDA in acetonitrile at rt. | ||||||
| 1 | DMSO | PIFA | 2 | rt | 1:1.4 |
55 |
| 2 | EtOAc | PIFA | 5 | rt | 1:1.1 |
72 |
| 3 | DMF | PIFA | 5 | rt | 1:1.4 |
85 |
| 4 | MeCN | PIFA | 4 | rt |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1.1 :1.1
|
87 |
| 5 | MeCN | PIDA | 3.5 | rt | 1:1.1 |
47 |
| 6 | MeCN | PIFA | 32 | 0 | 1:1.6 |
59 |
| 7 | MeCN | PIFA | 4 | 50 | 1:1.6 |
52 |
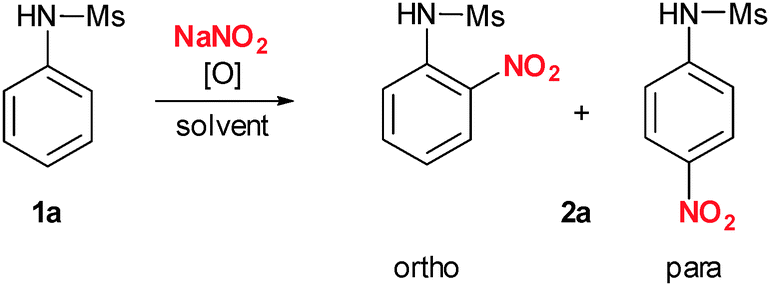
Changing the aryl-λ3-iodane from PIFA to PIDA (phenyl iodine diacetate) resulted in a significant lower yield of 47% (Table 1, entry 5). This observation is in good agreement with our proposal including a ligand-exchange as the initiating step since ligand exchange to generate A should be much faster with a less coordinating anion. Finally, lowering or increasing the temperature to 0 °C or 50 °C did also not result in improved product yields (Table 1, entries 6 and 7). Therefore, we decided to use the almost optimal reaction conditions described in entry 4 for our further systematic investigation of the substrate scope (Scheme 2). Electron-rich N-aryl sulfonamides bearing additional methoxy or alkyl substituents reacted well and in perfect regioselectivity. p-Methoxy and p-methyl derivatives 1b and 1d gave the corresponding o-nitro derivatives 2b and 2d in 62% and 67% yield whereas the o-methoxy substituted derivative 1c gave para selective nitration to yield 2c in 48%. Nitration of the 3,5-dimethyl substituted sulfonamide 1e resulted in formation of 2e in 60% yield. Interestingly, when phenyl substituted sulfonamide 1f was used, exclusive nitration of the N-aryl group of the sulfonamide was observed in 74% yield (2f). N-(1-Naphthyl) sulfonamide gave 2g in 66% yield in an almost 1:1 mixture of o/p regioisomers. Next, we investigated halogenated N-aryl sulfonamides. Chlorinated- and fluorinated derivatives reacted smoothly to give the desired products 2h–j in 66–79% yield. However, an additional iodine-substituent had a negative influence on reactivity resulting in formation of 2k in only 42% yield. Very electron-poor nitro-, cyano-, and ester-containing sulfonamides could be nitrated as well to give the desired products 2l–2p in 66–82% yield. Cyano- and chloro substituted sulfonamide 1q gave 2q in poor isolated yield of 24%. N-(3-Pyridyl) sulfonamide yielded the desired nitro-substituted pyridine derivative 2r in 56% yield. Finally, it is worth mentioning that N-unprotected aniline, N-methyl aniline and N,N-dimethylaniline gave messy reaction mixtures under similar reaction conditions.
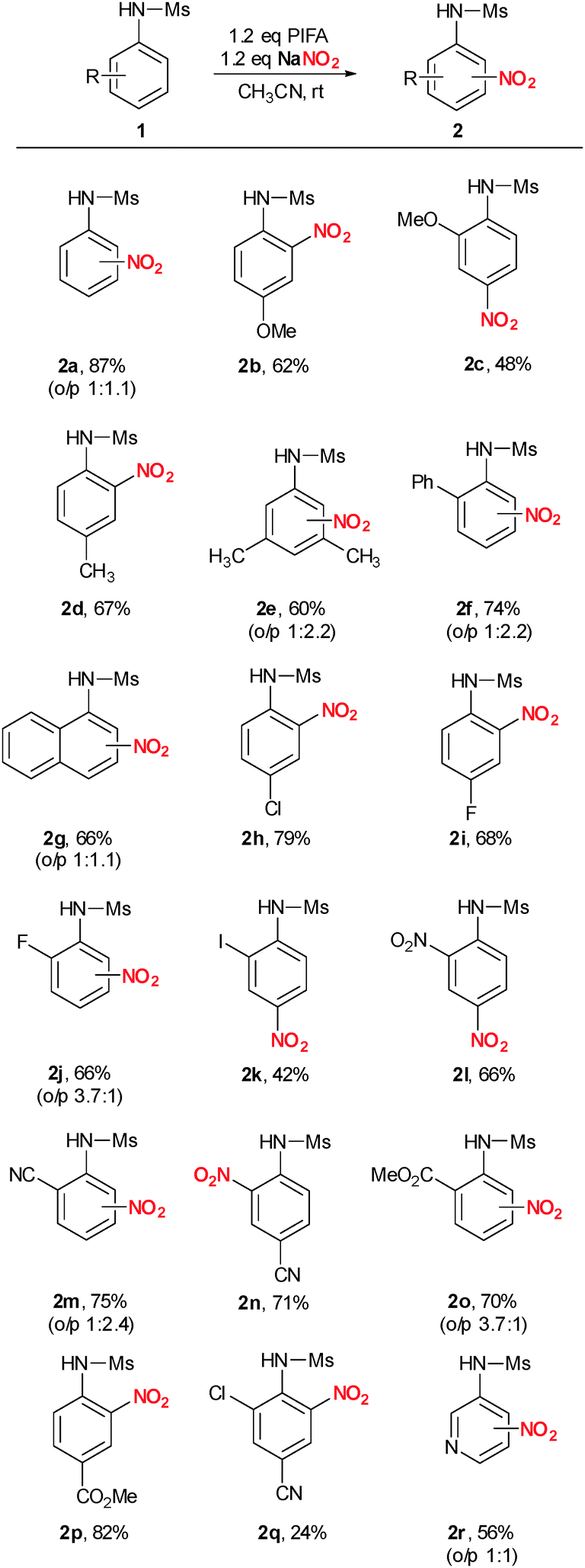 | ||
| Scheme 2 Substrate scope. Reaction conditions: 0.2 mmol 1a, 0.24 mmol NaNO2, 0.24 mmol PIFA in acetonitrile at rt. Reaction time: 1–48 h. | ||
To prove our initial proposal presented in Scheme 1, in particular the generation of a nitronium ion (Scheme 1a), a replacement experiment was performed (Scheme 3). When a mixture of 1a was treated with PIFA and NaNO2 in the presence of a slight excess of mesitylene 3, formation of 2a was observed exclusively in 59%. If a nitronium ion would be the active species in this transformation, the more electron-rich arene 3 should react significantly faster leading to significant amounts of 4. However, formation of 4 was not observed. In addition, we could not detect formation of the nitronium ion by Raman spectroscopy at a characteristic band at 1394 cm−1 when PIFA and NaNO2 were combined in acetonitrile.26
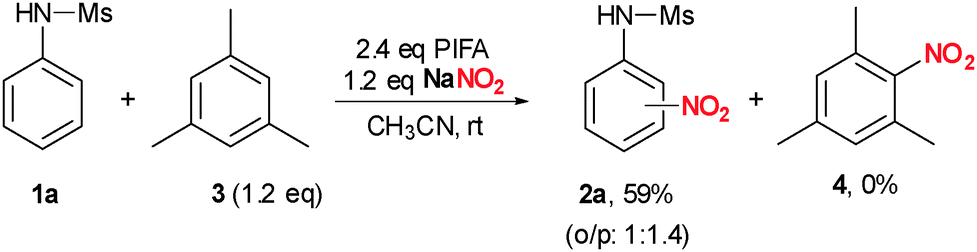 | ||
| Scheme 3 Replacement experiment. | ||
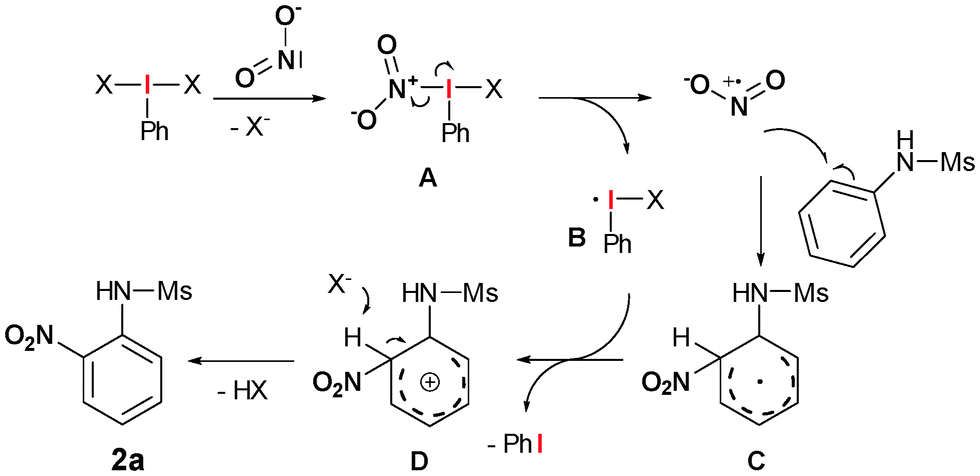
These experiments together with the observation that a brown gas with a characteristic pungent odour is formed at the beginning of the reaction, is a strong evidence for the oxidative formation of nitrogen dioxide via an 1-electron transfer-mediated decomposition of the aryl-λ3-iodane (Scheme 4).
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Initially, ligand exchange of the nitrite anion at the hypervalent iodine atom should give intermediate A. The labile N–I-bond subsequently decomposes to give nitrogen dioxide and the iodine-centred radical B. A radical reaction between NO2 and the arene gives a stabilized cyclohexadienyl radical C. Formation of this radical should be strongly stabilized by heteroatom containing functional groups which might be the reason for the high chemoselectivity of this transformation towards N-aryl sulphonamides. However, C can subsequently be oxidized by B to the corresponding cyclohexadienyl cation D. Final proton elimination yields the desired nitro arene 2a.
In conclusion we described the first hypervalent iodine-mediated nitration of electron deficient N-aryl sulfonamides. By using a combination of sodium nitrite as a remarkable cheap and easy to handle nitro source and the aryl-λ3-iodane PIFA as a mild oxidant the desired nitro (hetero)arenes could be isolated in up to 87% yield. This novel nitration method is acid free and subsequently tolerates a variety of additional functional groups on the (hetero)arene moiety. Based on these initial results we want to use the combination of a hypervalent iodine reagent and nitrite salts in the near future to introduce nitro functionalities into other, less reactive substrates in an oxidative manner.
We gratefully acknowledge the DFG (NA 955/1-1) and the Fonds der Chemischen Industrie (Sachkostenzuschuss) for financial support.
Notes and references
- N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, Inc., 2002, pp. 3–29 Search PubMed.
- G. A. Olah, R. Malhotra and S. C. Narang, Nitration Methods and Mechanisms, VCH Publishing, 2002 Search PubMed.
- G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554 CAS.
- D. Koley, O. C. Colón and S. N. Savinov, Org. Lett., 2009, 11, 4172 CrossRef CAS PubMed.
- Y. Liu, Synlett, 2011, 1184 CrossRef CAS PubMed.
- M. S. Kumar, K. C. Rajanna, K. R. Reddy, M. Venkateswarlu and P. Venkanna, Synth. Commun., 2013, 43, 2672 CrossRef CAS.
- T. Taniguchi, A. Yajima and H. Ishibashi, Adv. Synth. Catal., 2011, 353, 2643 CrossRef CAS.
- D. Hirose and T. Taniguchi, Beilstein J. Org. Chem., 2013, 9, 1713 CrossRef PubMed.
- T. Taniguchi, Y. Sugiura, T. Hatta, A. Yajima and H. Ishibashi, Chem. Commun., 2013, 49, 2198 RSC.
- S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Chem. Commun., 2013, 49, 5286 RSC.
- S. Maity, T. Naveen, U. Sharma and D. Maiti, Org. Lett., 2013, 15, 3384 CrossRef CAS PubMed.
- B. V. Rokade and K. R. Prabhu, Org. Biomol. Chem., 2013, 11, 6713 CAS.
- T. Shen, Y. Yuan and N. Jiao, Chem. Commun., 2014, 50, 554 RSC.
- B. Kilpatrick, M. Heller and S. Arns, Chem. Commun., 2013, 49, 514 RSC.
- Bretherick's handbook of reactive chemical hazards, Elsevier, Amsterdam, 2007 Search PubMed.
- A. R. Pourali and A. Goli, J. Chem. Sci., 2011, 123, 63 CrossRef CAS.
- T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS PubMed.
- V. V. Zhdankin, ARKIVOC, 2009, 1 CrossRef CAS.
- L. Pouysegu, T. Sylla, T. Garnier, L. B. Rojas, J. Charris, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 5908 CrossRef CAS PubMed.
- E. A. Merritt and B. Olofsson, Synthesis, 2011, 517 CAS.
- T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Tetrahedron, 2009, 65, 10797 CrossRef CAS PubMed.
- R. Samanta, J. O. Bauer, C. Strohmann and A. P. Antonchick, Org. Lett., 2012, 14, 5518 CrossRef CAS PubMed.
- R. Samanta, J. Lategahn and A. P. Antonchick, Chem. Commun., 2012, 48, 3194 RSC.
- A. P. Antonchick, R. Samanta, K. Kulikov and J. Lategahn, Angew. Chem., Int. Ed., 2011, 50, 8605 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed.
- H. G. M. Edwards and V. Fawcett, J. Mol. Struct., 1994, 326, 131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc04738a |
| This journal is © The Royal Society of Chemistry 2014 |