
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient labelling of enzymatically synthesized vinyl-modified DNA by an inverse-electron-demand Diels–Alder reaction†
Holger
Bußkamp
,
Ellen
Batroff
,
Andrea
Niederwieser
,
Obadah S.
Abdel-Rahman
,
Rainer F.
Winter
,
Valentin
Wittmann
and
Andreas
Marx
*
Department of Chemistry and Konstanz Research School Chemical Biology, University of Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany. E-mail: andreas.marx@uni-konstanz.de
First published on 22nd July 2014
Abstract
Many applications in biotechnology and molecular biology rely on modified nucleotides. Here, we present an approach for the postsynthetic labelling of enzymatically synthesized vinyl-modified DNA by Diels–Alder reaction with inverse electron demand using a tetrazine. Labelling proceeds very efficiently and supersedes several known approaches.
Conjugation of biopolymers with functional molecules, e.g. affinity tags or fluorescent dyes, is crucial for many applications in molecular biology.1 Conjugation of a biopolymer with a label is often a very challenging task. Bioorthogonal ligation reactions have been reported, which enable the site specific labelling of modified biopolymers, such as DNA.2 For example, copper-catalyzed alkyne azide cycloaddition,3 Staudinger ligation,4 Diels–Alder reaction,5 Michael addition,6 reductive amination7 and Suzuki cross coupling8 have successfully been applied to modify DNA. These labelling reactions have in common that the DNA and the label have to bear suitable reactive groups, which are used for conjugation. The introduction of these reactive moieties into DNA can also be challenging. In nature, DNA is synthesized by DNA polymerases, which catalyze the addition of nucleoside monophosphates to the 3′-end of an existing DNA polymer by using nucleoside triphosphates. Polymerases are also able to incorporate modified nucleotide analogues.9 Therefore it is possible to introduce reactive moieties in DNA, which can afterwards be exploited for bioorthogonal conjugation. For the labelling of DNA the Diels–Alder reaction is of particular interest. It capitalizes on the selective cycloaddition of alkenes and dienes and does not require catalysts or toxic reagents. For Diels–Alder reactions with inverse electron demand (DARinv) 1,2,4,5-tetrazines5b have been used with strained alkenes, such as trans-cyclooctenes,10 norbornenes5f,11 or cyclopropenes,12 resulting dihydropyridazine derivatives. Furthermore, it was shown that tetrazines also react with terminal alkenes.13
For the postsynthetic modification of enzymatically synthesized DNA via DARinv (Fig. 1A) we envisioned 7-vinyl-7-deaza-2′-deoxyadenosine (dvinA) and 5-vinyl-2′-deoxyuridine (dvinU) (Fig. 1B) as nucleoside analogues bearing the vinyl group at the 7 or 5 position, respectively. The vinyl group is a very small modification that might render nucleosides into dienophiles for labelling by DARinv. In both cases the reactive group points towards the major groove of the DNA duplex and thus should be accessible by labelling reagents. The nucleosides were synthesized according to known procedures.14 As the reaction partner in the Diels–Alder reaction a known biotinylated tetrazine derivative (Fig. 1B) was used.13
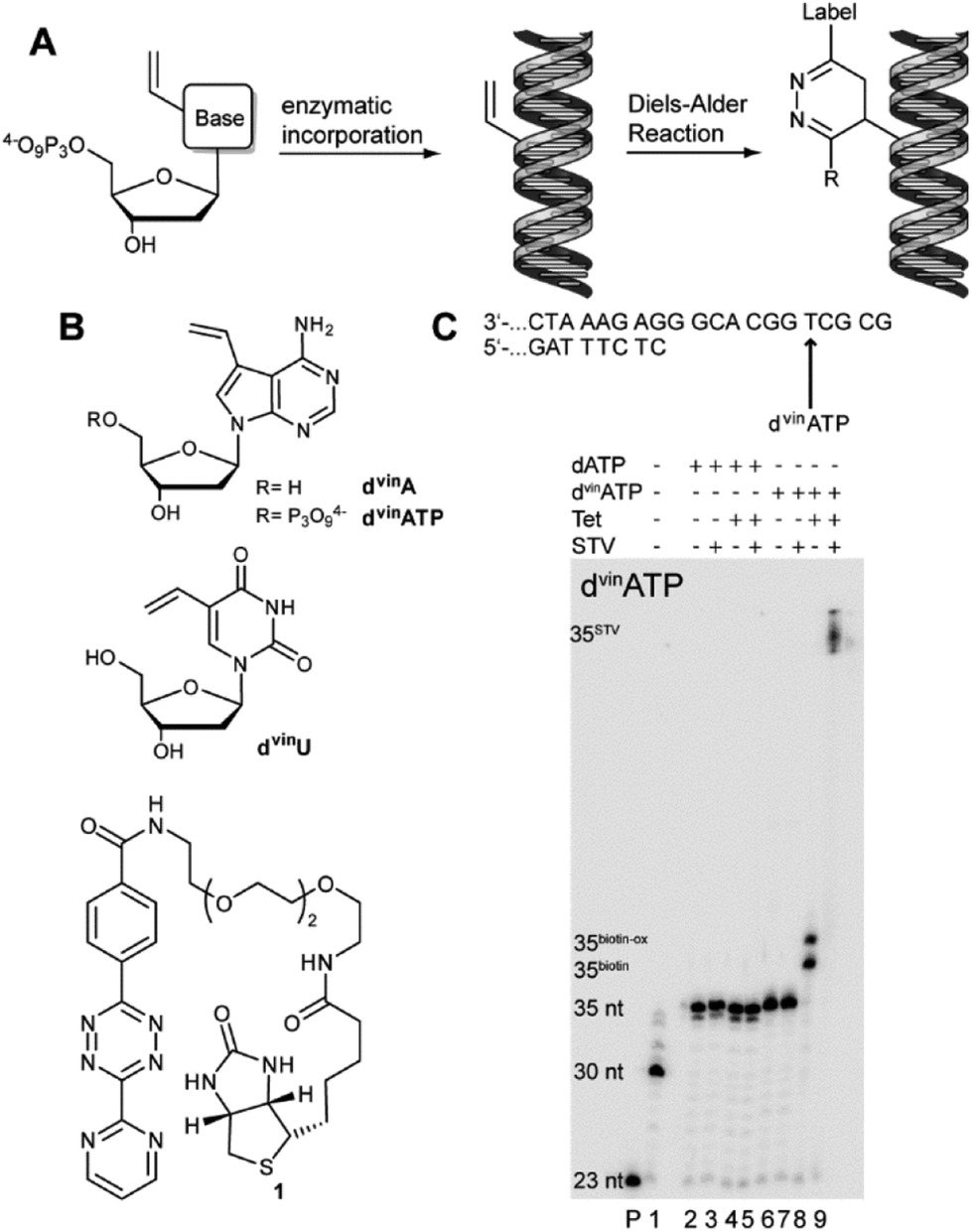 | ||
| Fig. 1 (A) Schematic representation of the labelling strategy. Vinyl-modified dNTP-analogues are enzymatically incorporated into the DNA duplex. The biopolymer is then subjected to DARinv with a tetrazine-modified label. (B) Structures of dvinA, dvinATP, dvinU and tetrazine 1. (C) Top: a partial primer template complex used for the primer extension studies. Incorporation position for dvinATP is indicated by an arrow. PAGE analysis of PEX and DARinv with dvinATP. Tet: incubation with tetrazine 4; STV: incubation with streptavidin; P: primer only; 1: PEX by KlenTaq DNA polymerase in the presence of dCTP, dGTP and dTTP; 2: all four natural dNTPs; 3: the same as 2 but followed by incubation with streptavidin (STV); 4: the same as 2 but followed by incubation with tetrazine 1; 5: the same as 4 but followed by incubation with STV; 6: the same as 1 but in the presence of dvinATP; 7: the same as 6 but followed by incubation with STV; 8: the same as 6 but followed by incubation with tetrazine 1; and 9: the same as 8 but followed by incubation with STV. | ||
To test the ability of the nucleosides to undergo DARinv with tetrazines, we determined the second-order rate constants k2 (Fig. S1, ESI†). dvinA undergoes DARinv with a constant k2 of 0.140 (±0.001) (L mol−1 s−1) and dvinU with a rate constant k2 of 0.010 (±0.0002) (L mol−1 s−1). The kinetic rate constant of dvinU is comparable to a recent report.15 We focused on dvinA in this study, as the DARinv proceeds 14-fold faster than the reaction with dvinU. Furthermore, the rate constant of dvinA is about 10-fold higher than that of the reaction of terminal alkenes with tetrazines, which has been reported for efficient labelling.13 In comparison to other prominent labelling reactions, the DARinv using dvinA proceeds faster than the Staudinger ligation16 or the strain-promoted azide–alkyne cycloaddition of dibenzocyclooctyne.17 Thus, labelling with dvinA and tetrazines appears to be promising for further applications.
The differences in the reactivity of the nucleobases might be explained by their different electronic properties. To gain further insights, we calculated the frontier orbitals of the tetrazine and the vinylated nucleobases and evaluated the energy difference of the interacting orbitals as an estimate for the reactivity.18 We found that the energy difference between the interacting orbitals (LUMO + 1 of the tetrazine, HOMO of the nucleobases; Fig. S2, ESI†) of the tetrazine and dvinU is larger by 0.55 eV than the difference between the corresponding orbitals when considering dvinA. The smaller energy difference can explain the higher reactivity of dvinA in comparison to dvinU.
Encouraged by these results, we synthesized dvinATP by phosphorylation as described in ref. 19. We tested the acceptance of the nucleotide analogue by KlenTaq DNA polymerase and conducted primer extension reactions (PEX) using a radioactively labeled primer strand. We employed standard conditions for primer extension reactions as they have been reported before.9a,b The nucleotide analogue was incorporated efficiently into the DNA strand. A full length product was observed in all cases and no stalling of the polymerase during incorporation of the modified nucleotide took place under the employed conditions, indicating efficient processing of the modified building block by the DNA polymerase.
Next, we tested if the incorporated modification can engage in DARinv in double stranded DNA (dsDNA). The primer extension product was incubated with tetrazine 1, bearing a biotin moiety. The DNA was used in low concentrations (0.9 μM) while an excess of labelling reagent (1.7 mM) was used. After purification via gel filtration, an aliquot was incubated with streptavidin. All samples were analyzed by denaturing polyacrylamide gel electrophoresis (PAGE). dvinA embedded in dsDNA undergoes DARinv with the biotinylated tetrazine derivative 1, indicated by the change in the migration of the product by PAGE analysis (Fig. 1C). Further control reactions point out that only when dvinATP was used as the substrate and dvinA was incorporated, the large shift of the product migration was detected by PAGE analysis after subsequent incubation with biotinylated tetrazine 1 and streptavidin.
Next, we investigated the efficiency of the reaction by varying the reaction time (Fig. 2). Interestingly, the starting material was almost completely consumed within 10 minutes. During longer incubation times another band appears that might be derived from the oxidation of the Diels–Alder reaction product, as it is known that dihydropyridazines easily oxidize to pyridazines in the presence of oxygen.13
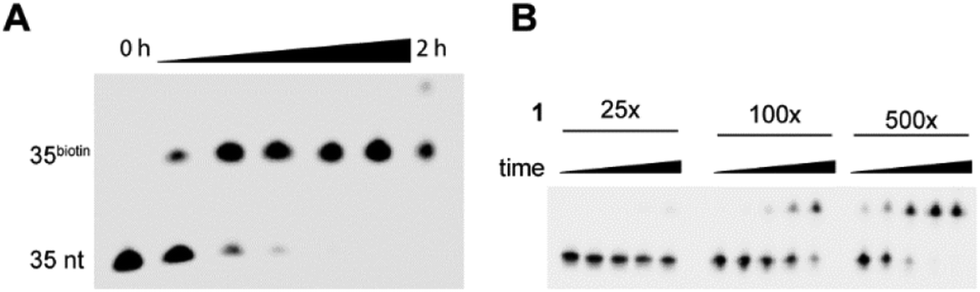 | ||
| Fig. 2 (A) A time course experiment with 1.7 mM tetrazine (samples taken after 0 min, 5 min, 10 min, 20 min, 30 min, 1 h and 2 h). After 10 minutes almost all of the starting material is consumed. (B) Effect of the excess of tetrazine on the reaction speed. 25-(25×, 22 μM) to 500-fold excess (500×, 450 μM) of tetrazine was added. The reactions were stopped after different time points (5 min, 10 min, 30 min, 1 h, and 2 h). | ||
Next, we investigated the dependence of the reaction efficiency on the concentration of the labelling reagent. Therefore, the product of the primer extension reaction in the presence of dvinATP was incubated with increasing concentrations of tetrazine 1. As an excess of tetrazine was applied, the reaction follows the kinetics of pseudo-first order. We stopped each of the reactions after different time points and analyzed the reactions by PAGE. Interestingly, when 22 μM labelling reagent is present, conversion to the reaction product occurred only to a minor extent even after prolonged reaction times (2 h). When 90 μM labelling reagent is used, after 2 hours about 70% of the starting material has been converted to the reaction product. Upon applying 450 μM tetrazine 1 about 70% conversion took place after 30 minutes and after 1 hour no starting material was detected anymore. To gain deeper insights, we performed kinetic studies of the DARinv on the DNA duplex (Fig. S3, ESI†). We estimated the second order rate constant k2 of 0.42 (±0.03) (L mol−1 s−1). This constant is 3-fold higher than for the nucleoside. This increase in the reaction rate might originate from the polar microenvironment in the major groove, in which the Diels–Alder reaction takes place, as it has been reported that Diels–Alder reactions are accelerated in polar solvents.20 Furthermore we found that even at low DNA concentration (0.9 μM), applying 450 μM of tetrazine results in very efficient labelling within 30 min. In comparison, 4 h are required for labelling by copper-catalyzed azide–alkyne cycloaddition in a similar setup when the same concentration of DNA albeit a 1900-fold excess of labelling reagent was used.9b
Next, we investigated if dvinATP can also be used in the polymerase chain reaction (PCR) by completely substituting natural dATP by dvinATP (Fig. 3). With the complete exchange of natural dATP by dvinATP, the full length product could be observed. The incorporation of 160 modified nucleotides into the DNA duplex was indicated by a change in the migration behavior of the DNA product, as monitored by gel electrophoresis analysis.
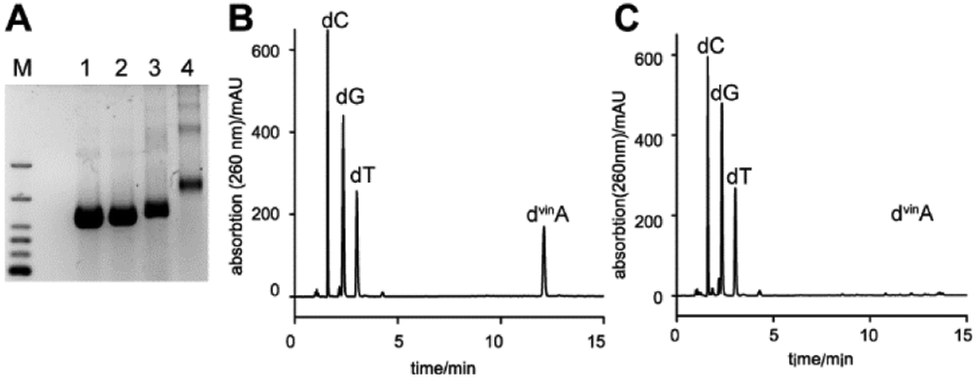 | ||
| Fig. 3 (A) PCR of a 414 bp DNA fragment with dATP completely exchanged by dvinATP. M: marker, 700 bp, 500 bp, 400 bp, 300 bp, 200 bp, and 100 bp. 1: PCR with natural dNTPs; 2: the same as 1 but incubation with 1.7 mM tetrazine 1 for 1 h at room temperature; 3: PCR with dATP exchanged completely by dvinATP; and 4: the same as 3 but with incubation of 1.7 mM tetrazine 1 for 1 h at room temperature. PCR reaction without any dATP yielded no product (data not shown). (B) HPLC chromatogram of the digested PCR product without incubation with tetrazine 1 and (C) HPLC chromatogram of the digested PCR product with incubation of tetrazine 1. | ||
Still, the PCR product containing the vinyl groups undergoes DARinv when incubated with tetrazine for 1 h at room temperature, visible in a large shift of the band after addition of 1 (Fig. 3). As only one major band appeared after Diels–Alder reaction, one can conclude that a defined reaction product is formed, indicating that in most DNA strands a similar number of vinyl groups reacted with tetrazine. To address the efficiency of the DARinv using the PCR product, we digested the PCR product to the nucleosides and analyzed the products by HPLC. We found that the peak in the HPLC chromatogram corresponding to dvinA completely vanished and a new peak appeared at higher retention times (Fig. S4, ESI†) when the PCR product was incubated with tetrazine 1. These findings indicate that all vinyl groups reacted with tetrazine.
To summarize, here we show that the modified nucleoside triphosphate dvinATP is efficiently incorporated into DNA by a DNA polymerase. The modified biopolymer can undergo DARinv and can therefore be conjugated. We found that natural dATP can be substituted completely by its modified counterpart in PCR and still the full length product is formed, which can react in DARinv. All in all, we present an efficient tool for labelling and manipulation of enzymatically synthesized DNA.
We gratefully acknowledge funding by the DFG (SFB 969) and the Konstanz Research School Chemical Biology.
Notes and references
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13 CrossRef CAS PubMed.
- S. H. Weisbrod and A. Marx, Chem. Commun., 2008, 5675 RSC.
- (a) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC; (b) P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350 CrossRef CAS PubMed; (c) P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442 CrossRef CAS PubMed; (d) F. Seela, V. R. Sirivolu and P. Chittepu, Bioconjugate Chem., 2007, 19, 211 CrossRef PubMed.
- (a) H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635 CrossRef CAS; (b) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS; (c) S. H. Weisbrod and A. Marx, Chem. Commun., 2007, 1828 RSC.
- (a) O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS; (b) J. Sauer, et al. , Eur. J. Org. Chem., 1998, 2885 CrossRef CAS; (c) M. Hocek, Eur. J. Org. Chem., 2003, 245 CrossRef CAS; (d) V. Borsenberger and S. Howorka, Nucleic Acids Res., 2009, 37, 1477 CrossRef CAS PubMed; (e) J. Schoch, M. Wiessler and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8846 CrossRef CAS PubMed; (f) J. Schoch and A. Jäschke, RSC Adv., 2013, 3, 4181 RSC; (g) J. Schoch, et al. , Bioconjugate Chem., 2012, 23, 1382 CrossRef CAS PubMed.
- J. Dadová, et al. , Angew. Chem., Int. Ed., 2013, 52, 10515 CrossRef PubMed.
- V. Raindlová, R. Pohl and M. Hocek, Chem. – Eur. J., 2012, 18, 4080 CrossRef PubMed.
- L. Lercher, et al. , Angew. Chem., Int. Ed., 2013, 52, 10553 CrossRef CAS PubMed.
- (a) S. Obeid, et al. , J. Am. Chem. Soc., 2013, 135, 15667 CrossRef CAS PubMed; (b) S. Obeid, et al. , Chem. Commun., 2012, 48, 8320 RSC; (c) S. Jäger, et al. , J. Am. Chem. Soc., 2005, 127, 15071 CrossRef PubMed.
- (a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef CAS PubMed; (b) N. K. Devaraj, et al. , Angew. Chem., Int. Ed., 2010, 49, 2869 CrossRef CAS PubMed; (c) R. Rossin, et al. , Angew. Chem., Int. Ed., 2010, 49, 3375 CrossRef CAS PubMed.
- (a) T. Plass, et al. , Angew. Chem., Int. Ed., 2012, 51, 4166 CrossRef CAS PubMed; (b) H. S. G. Beckmann, A. Niederwieser, M. Wiessler and V. Wittmann, Chem. – Eur. J., 2012, 18, 6548 CrossRef CAS PubMed.
- (a) A.-K. Späte, et al. , Bioconjugate Chem., 2013, 25, 147 CrossRef PubMed; (b) D. M. Patterson, et al. , J. Am. Chem. Soc., 2012, 134, 18638 CrossRef CAS PubMed; (c) J. Yang, J. Šečkutė, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- A. Niederwieser, et al. , Angew. Chem., Int. Ed., 2013, 52, 4265 CrossRef CAS PubMed.
- F. Seela, M. Zulauf, M. Sauer and M. Deimel, Helv. Chim. Acta, 2000, 83, 910 CrossRef CAS.
- U. Rieder and N. W. Luedtke, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201403580.
- F. L. Lin, et al. , J. Am. Chem. Soc., 2005, 127, 2686 CrossRef CAS PubMed.
- N. E. Mbua, et al. , ChemBioChem, 2011, 12, 1912 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC.
- L. Ötvös, J. Sági, T. Kovács and R. T. Walker, Nucleic Acids Res., 1987, 15, 1763 CrossRef PubMed.
- J. W. Wijnen, S. Zavarise, J. B. F. N. Engberts and M. Charton, J. Org. Chem., 1996, 61, 2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details of dvinATP and experimental details of kinetic measurements, PEX, PCR, labelling with tetrazine, and the protocol of the digestion of the PCR product to nucleosides. See DOI: 10.1039/c4cc04332d |
| This journal is © The Royal Society of Chemistry 2014 |