 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Merging of inner and outer ruthenium organometallic coordination motifs within an azuliporphyrin framework†
Michał J.
Białek
and
Lechosław
Latos-Grażyński
*
Department of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: lechoslaw.latos-grazynski@chem.uni.wroc.pl
First published on 30th June 2014
Abstract
The insertion of ruthenium(II) into an azuliporphyrin (TPAP) has yielded carbonyl ruthenium(II) azuliporphyrin [Ru(TPAP)(CO)] featuring an equatorial CNNN set of donors. Its azulene moiety serves as the π-coordination platform to accommodate the Ru4(CO)9 cluster. This chemistry proved to be general giving rise to a series of bimetallic complexes [M(TPAP){Ru4(CO)9}] (M = Ru(CO), Ni, Pd, Pt).
The dynamic development of synthetic routes for carbaporphyrinoids allows for the exploration of porphyrin-like or porphyrin-unlike coordination chemistry.1–3 The scope of possible coordination modes includes metal–carbon (sp2) and metal–carbon (sp3) σ-bonds, an agostic side-on interaction between the inner C–H and a metal centre, and an intramolecular metal(II)–η2–CC interaction inside a porphyrinoid frame. A unique π-coordination was identified in metalloceneporphyrinoids.4 More intricate organometallic structures have been reported which include the tripalladium sandwich complex consisting of two dianionic palladium(II) dicarbaporphyrin units surrounding a palladium(IV) cation,5 and a gable-porphyrin-type organization where two N-confused porphyrins are bridged by the rhodium Rh4(CO)4 cluster.6
Unconventional coordination motifs can be foreseen presuming that the π-surface of carbaporphyrinoids provides several potential metal binding sites by analogy to the π-complexes reported for tetrapyrrolic ligands where the external coordination involves pyrrole fragments.7
Facile introduction of an azulene fragment instead of one pyrrolic unit gives an azuliporphyrin8,9 – a highly attractive candidate for exploration of organometallic chemistry inside a carbaporphyrinoid core.1 Until now, meso-tetraaryl and β-alkylated azuliporphyrins have formed organometallic compounds where a direct M–C σ-bond [M = Ni(II), Pd(II), Pt(II), Ir(III)] has been solely detected.2,10–12 This study aims to explore the diversity of ruthenium toward azuliporphyrin coordination modes.
A ruthenium(II) ion has been readily inserted into azuliporphyrin 1, following the procedure reported previously for ruthenium(II) regular porphyrins, to give the corresponding complexes 2 and 3 in which the azuliporphyrin acts as a dianionic organometallic ligand (Scheme 1).
 | ||
| Scheme 1 Reaction of ruthenium dodecacarbonyl with an azuliporphyrin. | ||
The regular complex 2 can be detected and isolated when 1 is reacted with a sub- or equimolar amount of [Ru3(CO)12]. In the presence of a ruthenium source in excess, only the cluster complex 3 is obtained (see ESI†). This step reveals the unique coordination reactivity of the azulene unit, merging two (inner and perimeter) coordination motifs. Namely, the ruthenium(II) azuliporphyrin binds the Ru4(CO)9 cluster taking advantage of the azulene π-surface.
Observations of reactivity of compound 3 show essentially three kinds of such toward some nucleophilic agents (deactivated alumina, pyridine, acetonitrile), i.e. contraction of a seven-membered ring leading to demetalated benzocarbaporphyrins, secondary cluster reactivity connected with its stepwise substitution and complete cluster removal forming 2.
The effects of π-coordination are highly pronounced in spectroscopy as well as in the solid state. The 1H NMR spectra of the tripyrrolic unit, which is common for structures 2 and 3 (Fig. 1), resemble the basic pattern of the azuliporphyrin consistent with its borderline aromaticity.8,9 The typical spectrum of 2 affords an AB spin system at δ = 7.70, H(7,18), and δ 7.62 ppm, H(8,17), with a coupling constant (3J = 4.9 Hz) typical for a pyrrole ring in porphyrinoids, which is accompanied by a singlet at δ = 7.69 ppm from H(12,13) of the central pyrrole unit. The NMR resonances of an azulene unit have been assigned to 2, namely: 1H NMR – 6.80 H(21,31), 6.38 H(22,32), 6.64 H(23) ppm; 13C NMR – 131.5 C(21,31), 136.9 C(22,32), 135.1 C(23) ppm.
 | ||
| Fig. 1 1H NMR spectra of (a) [Ru(TPAP)(CO)(Py-d5)] (2) 600 MHz, CDCl3, 270 K; (b) [Ru(TPAP)(CO){Ru4(CO)9}] (3) 600 MHz, CD2Cl2, 250 K. | ||
The remarkable relocations of 1H and 13C azulene resonances of 3 (1H NMR: 3.74 H(21,31), 4.84 H(22,32), 1.84 H(23) ppm; 13C NMR: 63.2 C(21,31), 66.7 C(22,32), 32.3 C(23) ppm) reflect a cluster attachment. Actually, the change in the azuliporphyrin coordination mode determined for 3 can be readily appreciated by an analysis of coordination shifts defined here as 1H or 13C chemical shift differences between 2 and 3 at chosen positions (Fig. 1). These values: 1H NMR – 3.06 H(21,31), 1.54 H(22,32), 4.80 H(23) ppm; 13C NMR – 68.3 C(21,31), 70.2 C(22,32), 102.5 C(23) ppm, clearly point toward the azulene moiety acting as a coordinating unit. Analogous coordination shifts were previously detected in the 1H NMR and 13C NMR spectra of the ruthenium cluster – regular azulene complexes.13,14 Coupling constants for azulene protons have also decreased from about 10 Hz, typical for azulene derivatives, to a range of 6–8 Hz.
Further investigation of metal(II) azuliporphyrin coordination abilities against ruthenium has led to a series of bimetallic complexes with the general formula [M(TPAP){Ru4(CO)9}], where M = Ni (4), Pd (5), Pt (6). Chemical shift patterns of these complexes resemble those of 3 (Fig. S3, ESI†), and the azulene unit proved to be an unambiguous spectroscopic probe which could be used to identify the perimeter binding.
The structure of azulene-uncoordinated regular complex 2 has been determined for two forms that differ in the sixth axial ligand: 4-(N,N-dimethylamine)pyridine – DMAP (2-1) or acetonitrile (2-2) (Fig. 2a and b). These two structures show the specific flexibility of the azulene fragment (Fig. 3). Thus, the azulene moiety is notably prone to conformational changes enforced by external stimuli. To describe the situation quantitatively, the value of the angle between the planes generated by the coordination centre (RuCNNN) and the tropylium ring of azulene can be compared. The deflection of azulene from the macrocyclic plane is significant and equals 15.73(13)° for 2-1 and 28.0(3)° for 2-2. This much higher deformation for structure 2-2 is consistent with the involvement of the seven-membered ring into strong C–H⋯π interactions as revealed by analysis of the Hirshfeld surface (Fig. S26, ESI†) as well as the higher deformation of the CNNN coordination centre (Table S2, ESI†). The discussed angle is even higher for structure 3 since it amounts to 43.3(3)°. This is the result of cluster-azulene interactions (Fig. 2c).
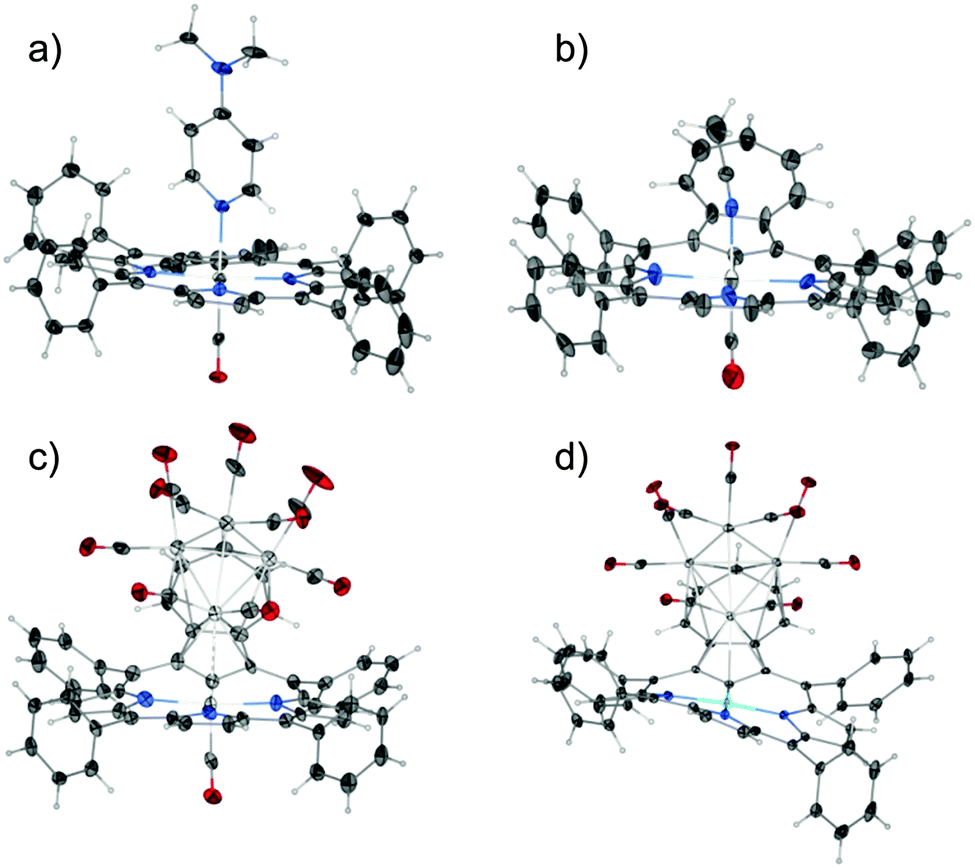 | ||
| Fig. 2 Presentation of the geometry of the complexes: (a) 2-1, (b) 2-2, (c) 3, (d) 4. Displacement ellipsoids drawn at 30% probability. | ||
 | ||
| Fig. 3 Ball-and-stick representations for fragments of 2-1, 2-2, 3 showing peculiar azulene flexibility. | ||
Compound 3 features an interesting metallic skeleton that can be described as a distorted tetrahedron (Ru2, Ru3, Ru4, Ru5) with close contact to the ruthenium(II) cation (Ru1) (Fig. 4a). Three out of the four ruthenium atoms (Ru2, Ru3, Ru4), which construct the tetrahedron skeleton, are directly coordinated to the azulene moiety and also bonded to two terminal carbonyl ligands.
 | ||
| Fig. 4 Cluster geometry and porphyrin conformations for (a) 3 and (b) 4. Phenyl rings removed to preserve clarity of the figure. | ||
The fourth ruthenium atom (Ru5) is linked to three basal ruthenium atoms and its coordination sphere is completed by three terminal carbonyl groups. To enable efficient facial coordination to the base of the tetrahedron the azulene moiety is strongly bent as described above.
In fact two modes of cluster coordination have been recognized. The five-membered ring of azulene prefers η5-coordination to one Ru2 atom in the standard half-sandwich fashion. The Ru4CO9 cluster coordinates all seven carbon atoms of the tropylium moiety (Fig. 2 and Fig. S28, ESI†). The five carbon atoms are bound to the other two ruthenium atoms in a η4-coordination mode, with the central carbon atom C(23) bonded to two ruthenium atoms. The fundamental features of the azulene–Ru4(CO)9 unit encompassed in 3 are reminiscent of the structure seen for the Ru4(CO)9 adduct to 1,3-bis(3-methylthienyl)azulene used as the fundamental building block in studies on post-coordination of multinuclear transition metal clusters to azulene-based polymers.15
In bimetallic complexes [Ni(TPAP){Ru4(CO)9}] 4, [Pd(TPAP){Ru4(CO)9}] 5, and [Pt(TPAP){Ru4(CO)9}] 6, the azulene moiety reveals the same coordination mode (Fig. S22–S24, ESI†). The deflection angle between the coordination core and the tropylium ring is 36.19(9)° and 32.8(2)°, for 4 and 5, respectively, and less pronounced for 6 – 24.69(15)° (Table S3, ESI†). The changes are significant in comparison to geometry of [Pt(TPAP)] (7) where this angle is 21.8(3)° and the azulene fragment is twisted in respect to a macrocyclic frame.
Azulene π-coordination results in the folding of a tripyrrolic brace (Fig. 2d and 4b). This can be expressed by the Cmeso–M–Cmeso angle between relevant Cmeso atoms and a central metal cation. The highest degree of folding can be noticed for 4, which results in C5–Ni1–C15 and C10–Ni1–C20 angles equal to 167.28(8)° and 168.24(9)°, respectively. This can be compared to the nearly planar [Pt(TPAP)] (7) (Fig. S25, ESI†), where the analogous angles are close to 180° (178.4(3)° and 178.4(2)°, respectively).
Within the tetrahedron of ruthenium atoms the Ru–Ru distances fall distinctly into two groups (Table S2, ESI†). In the first set Ru2–Ru4, Ru2–Ru5, Ru2–Ru3, Ru3–Ru4 distances vary from 2.8749(15) to 2.898(2) Å. The two other Ru–Ru distances are evidently shorter and amount to 2.714(2) (Ru3–Ru5) and 2.6857(16) Å (Ru4–Ru5). These values are within the limits of ruthenium–ruthenium bond lengths reported for a variety of ruthenium clusters, which diverge in the wide range of 2.65–3.20 Å (Fig. S29, ESI†). The suitable folding of azulene moiety forces the distinctive adjacency of the terminal Ru2 atom of the cluster and the azuliporphyrinic metal(II) cation. This distance equals 3.1598(18) Å in the case of Ru1–Ru2 and gradually grows in the series Ni1–Ru1 3.3113(5) Å, Pd1–Ru1 3.4074(12) Å, Pt1–Ru1 3.5883(16) Å.
As established by a detailed CSD database search (Fig. S29, ESI†) the Ru1–Ru2 value is within the limits of the longest distances considered for Ru–Ru bonding interactions detected in ruthenium clusters. The Ru2 atom definitely approaches the Ru1 cation at a distance much shorter than that expected for van der Waals contact. The Ru1–Ru2 distance of 3 can be compared with the longest bonding Ru–Ru contact detected in the first bis(metallabenzene) sandwich complex (bis(η6-3,5-dimethyl-1-tricarbonyl-ruthenabenzene)ruthenium) comprising two ruthenabenzene ligands whereas the Ru–Ru distance between the ruthenabenzene rings equals 3.38 Å.16 In terms of metal–metal bonded metalloporphyrins the structure of 3 provides an unprecedented structural motif as the sixth position is occupied by the ruthenium of the cluster, feasibly involved in a weak Ru1–Ru2 interaction. A distinct feature of the structure is the very pronounced bending of the Ru1–Ru2 axis from the perfect axial position (ca. 45°) as a consequence of strain induced by the incorporation of Ru2 into a Ru4(CO)9 scaffold.
The azulene moiety in the ruthenium(II) azuliporphyrin provides the suitable π-surface to bind the Ru4(CO)9 cluster. Two conceptually different organometallic motifs are merged in a unique three-dimensional architecture. We proved that these unique coordination abilities of azulene introduced into a carbaporphyrinoid frame are preserved in other suitable systems producing a series of bimetallic complexes [M(TPAP){Ru4(CO)9}], where M = Ni, Pd, Pt.
We thank Professor Tadeusz Lis for valuable discussions. Financial support from the NCN (Grant 2012/04/A/ST5/00593) is kindly acknowledged.
Notes and references
- M. Pawlicki and L. Latos-Grażyński, in Handbook of Porphyrin Science: with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, Singapore, 2010, ch. 8, pp. 103–192 Search PubMed.
- T. D. Lash, Chem. – Asian J., 2014, 9, 682 CrossRef CAS PubMed.
- A. Berlicka, P. Dutka, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2014, 53, 4885 CrossRef CAS PubMed.
- I. Grocka, L. Latos-Grażyński and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1044 CrossRef CAS PubMed.
- D. I. AbuSalim, G. M. Ferrence and T. D. Lash, J. Am. Chem. Soc., 2014, 136, 6763 CrossRef CAS PubMed.
- M. Toganoh, T. Niino, H. Maeda, B. Andrioletti and H. Furuta, Inorg. Chem., 2006, 45, 10428 CrossRef CAS PubMed.
- L. Cuesta and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 2716 RSC.
- T. D. Lash and S. T. Chaney, Angew. Chem., Int. Ed. Engl., 1997, 36, 839 CrossRef CAS.
- D. A. Colby and T. D. Lash, Chem. – Eur. J., 2002, 8, 5397 CrossRef CAS.
- S. R. Graham, G. M. Ferrence and T. D. Lash, Chem. Commun., 2002, 894 RSC.
- T. D. Lash, D. A. Colby, S. R. Graham, G. M. Ferrence and L. F. Szczepura, Inorg. Chem., 2003, 42, 7326 CrossRef CAS PubMed.
- T. D. Lash, K. Pokharel, M. Zeller and G. M. Ferrence, Chem. Commun., 2012, 48, 11793 RSC.
- Y. L. K. Tan and W. K. Leong, J. Organomet. Chem., 2011, 696, 2373 CrossRef CAS PubMed.
- K. Matsubara, K. Ryu, T. Maki, T. Iura and H. Nagashima, Organometallics, 2002, 21, 3023 CrossRef CAS.
- F. Wang, Y. H. Lai and M. Y. Han, Org. Lett., 2003, 5, 4791 CrossRef CAS PubMed.
- U. Effertz, U. Englert, F. Podewils, A. Salzer, T. Wagner and M. Kaupp, Organometallics, 2003, 22, 264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, figures of spectra (1H, 13C NMR; HRMS; UV-Vis), detailed crystallographic description and data. CCDC 1006123–1006129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc04271a |
| This journal is © The Royal Society of Chemistry 2014 |