 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A modular tyrosine kinase deoxyribozyme with discrete aptamer and catalyst domains†
Victor
Dokukin
and
Scott K.
Silverman
*
Department of Chemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, IL 61801, USA. E-mail: scott@scs.illinois.edu
First published on 30th June 2014
Abstract
We assess the utility of integrating a predetermined aptamer DNA module adjacent to a random catalytic DNA region for identifying new deoxyribozymes by in vitro selection. By placing a known ATP aptamer next to an N40 random region, an explicitly modular DNA catalyst for tyrosine side chain phosphorylation is identified. The results have implications for broader identification of deoxyribozymes that function with small-molecule substrates.
Deoxyribozymes are specific DNA sequences with catalytic activity.1 All known deoxyribozymes have been identified by in vitro selection2 from random-sequence DNA pools. For deoxyribozymes that function with oligonucleotide substrates (e.g., for RNA cleavage3), much of the catalyst–substrate binding interaction—and therefore the corresponding binding energy—is provided by Watson–Crick contacts, which are straightforwardly preprogrammed. These preprogrammed binding contacts enable the in vitro selection process to focus upon finding initially random DNA sequences that are tasked primarily with catalysis. However, for small-molecule substrates that inherently cannot engage in extensive Watson–Crick interactions, the initially random DNA region (e.g., N40) must instead simultaneously bind the substrate and catalyze the desired reaction, rather than solely participate in catalysis (Fig. 1A). Therefore, here we consider an explicitly modular approach to deoxyribozyme catalysis, in which a predefined aptamer (binding) domain engages in aptamer-like non-Watson–Crick contacts with the small-molecule substrate, allowing a distinct “catalytic” (enzyme) domain, subsequently identified through in vitro selection in the presence of the aptamer domain, to be devoted to catalysis (Fig. 1B). Many natural protein enzymes are functionally modular,4 and the group I intron ribozyme is modular in binding its RNA substrate.5 The present study had two major goals. First, we wished to determine whether providing a predefined aptamer module can lead to deoxyribozymes that make functional use of this module during catalysis. Second, we sought to assess whether the availability of the aptamer module provides more robust access by in vitro selection to new deoxyribozymes for a particular chemical reaction (e.g., leads to more numerous or faster DNA catalysts).
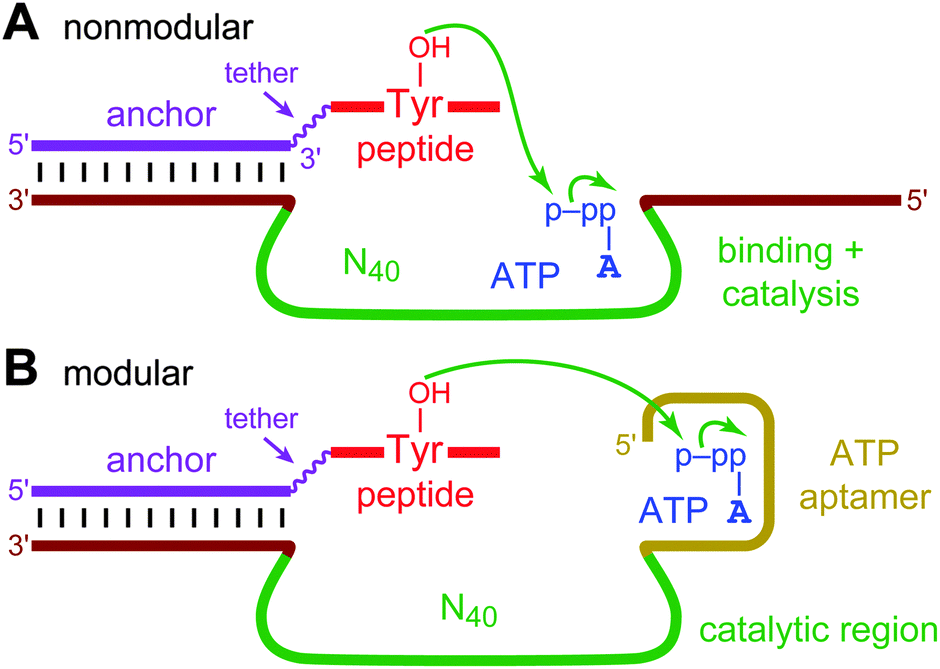 | ||
| Fig. 1 Nonmodular and modular deoxyribozymes, illustrated with tyrosine kinase activity for a DNA-anchored peptide substrate and ATP as the phosphoryl donor. (A) A nonmodular DNA catalyst, for which the initially random DNA region (e.g., N40; green) both binds the small-molecule substrate and performs catalysis. The two fixed sequences (brown) are required for PCR amplification during selection. The fixed sequence on the 5′-side of the random region can also contribute to substrate binding and catalysis. (B) A modular DNA catalyst. A predefined ATP aptamer region (gold) is located adjacent to the initially random region, which is responsible for catalysis. | ||
To achieve these goals, we used a known 27-nucleotide Mg2+-dependent ATP DNA aptamer domain6 that has been structurally characterized7 and is the basis for a wide range of experiments that involve sensors8 or other applications.9 Here, this ATP aptamer was integrated near N30, N40, and N50 DNA random regions, and in vitro selection was performed for tyrosine kinase activity towards a DNA-anchored CAAYAA hexapeptide substrate. The hexapeptide was connected to the DNA anchor by a hexa(ethylene glycol), or HEG, tether. Kinase activity by nucleic acid enzymes has been studied extensively for oligonucleotide substrates.10 We recently reported the first tyrosine kinase deoxyribozymes, which phosphorylate the tyrosine side chain of a peptide substrate and were identified from N30–N50 random regions that lack any predefined aptamer domain.11 Our current experiments were performed analogously, using the previously identified 8VP1 deoxyribozyme12 as the “capture” catalyst to enable the selection process (Fig. 2). The incubation conditions during the key selection step were 70 mM HEPES, pH 7.5, 1 mM ZnCl2, 20 mM MnCl2, 40 mM MgCl2, 150 mM NaCl, and 30 μM ATP at 37 °C for 14 h. Because the Kd for the isolated ATP aptamer is on the order of 6 μM,6 30 μM ATP is likely saturating while unlikely to induce the catalytic suppression that we observed at much higher NTP concentrations in our previous effort with nonmodular deoxyribozymes (these DNA catalysts used either 5′-triphosphorylated RNA or GTP as the phosphoryl donor).11 The 8VP1 capture control reaction typically had 25–35% yield in each selection round, which sets a corresponding upper limit on the observable yield from each random pool.
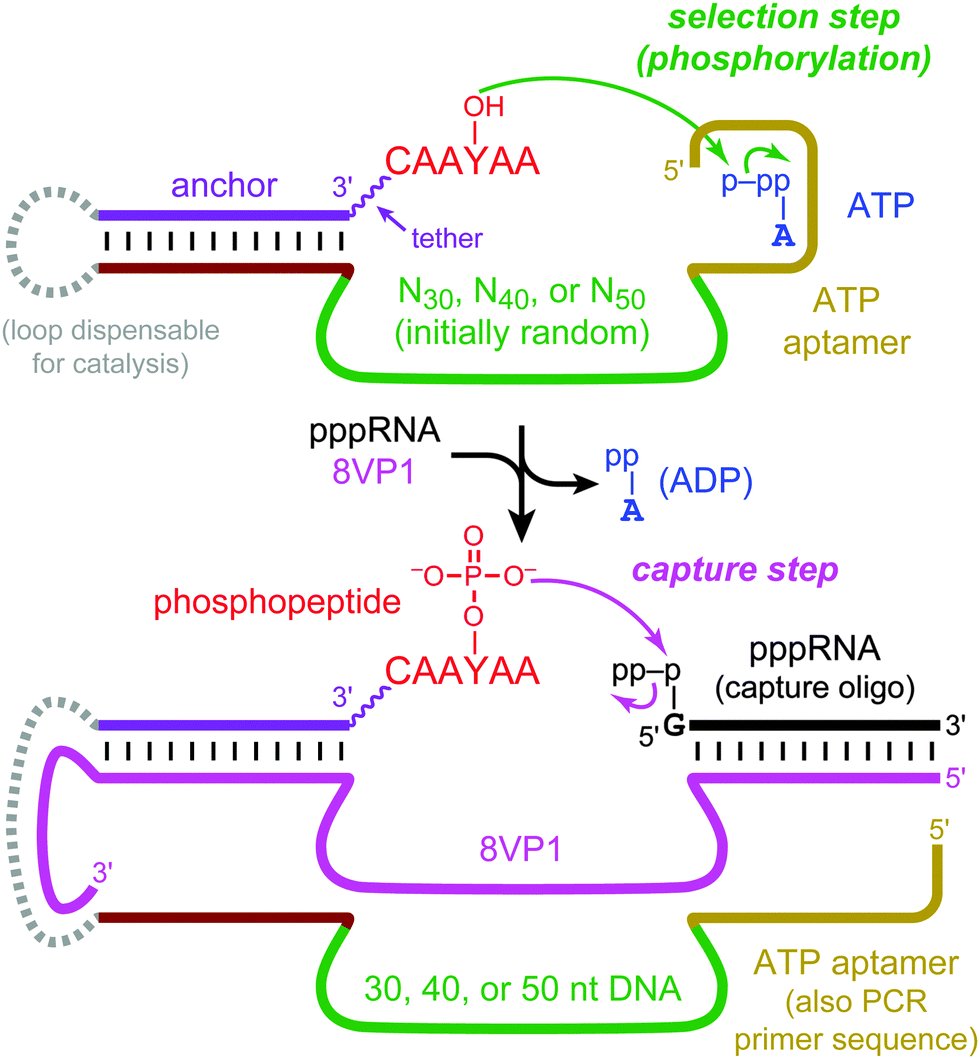 | ||
| Fig. 2 In vitro selection of modular tyrosine kinase deoxyribozymes. DNA-catalyzed peptide phosphorylation is followed by “capture” using the phosphotyrosine-specific 8VP1 deoxyribozyme and a 5′-triphosphorylated RNA oligonucleotide, enabling PAGE-shift separation of kinase deoxyribozymes. The capture step is followed by PCR (not shown) to initiate the next selection round. See Fig. S1, ESI† for the full chemical structure of the DNA-anchored peptide substrate and Fig. S2, ESI† for all nucleotide details. | ||
After 13 rounds, the N40 selection showed 0.9% yield, which rose to 3.1% at round 14 and 6.9% at round 17 (Fig. S3, ESI;† all of these yield values should be compared to the 25–35% maximum capture yield as indicated above). In contrast, neither the N30 nor the N50 selection gave more than 0.7% yield through round 18. The round 14 N40 pool was cloned and sequenced, revealing that the population had converged on a single active sequence named 14JS101.‡ The intact ATP aptamer domain was necessarily present as part of the 14JS101 sequence, as enforced by the method of selection (the forward PCR primer used to form the deoxyribozyme pool at the end of each selection round is the ATP aptamer sequence; Fig. 2). The 14JS101 deoxyribozyme provided up to 28% phosphorylation yield in 16 h when evaluated under the same incubation conditions as were used during the selection process, in a single-turnover assay with the DNA-anchored CAAYAA substrate provided separately from the deoxyribozyme. Note that this assay, which was performed as described,11 directly monitors conversion of YOH → YP; no capture step is involved. Zn2+ was strictly required for 14JS101 catalysis, with an optimum yield at 1.0 mM; omitting either Mn2+ or Mg2+ led to a 36-fold and eight-fold decrease in yield, respectively (Fig. S4, ESI†). The phosphorylation yield of 14JS101 was optimized to 52% by incubation in 40 mM Tris, pH 7.5, 1 mM ZnCl2, 10 mM MnCl2, 40 mM MgCl2, 150 mM NaCl, and 30 μM ATP at 37 °C (Fig. 3A). These optimized conditions were adopted for all further assays. The identity of the DNA-anchored hexapeptide phosphorylation product was validated by MALDI mass spectrometry (Fig. S5, ESI†).
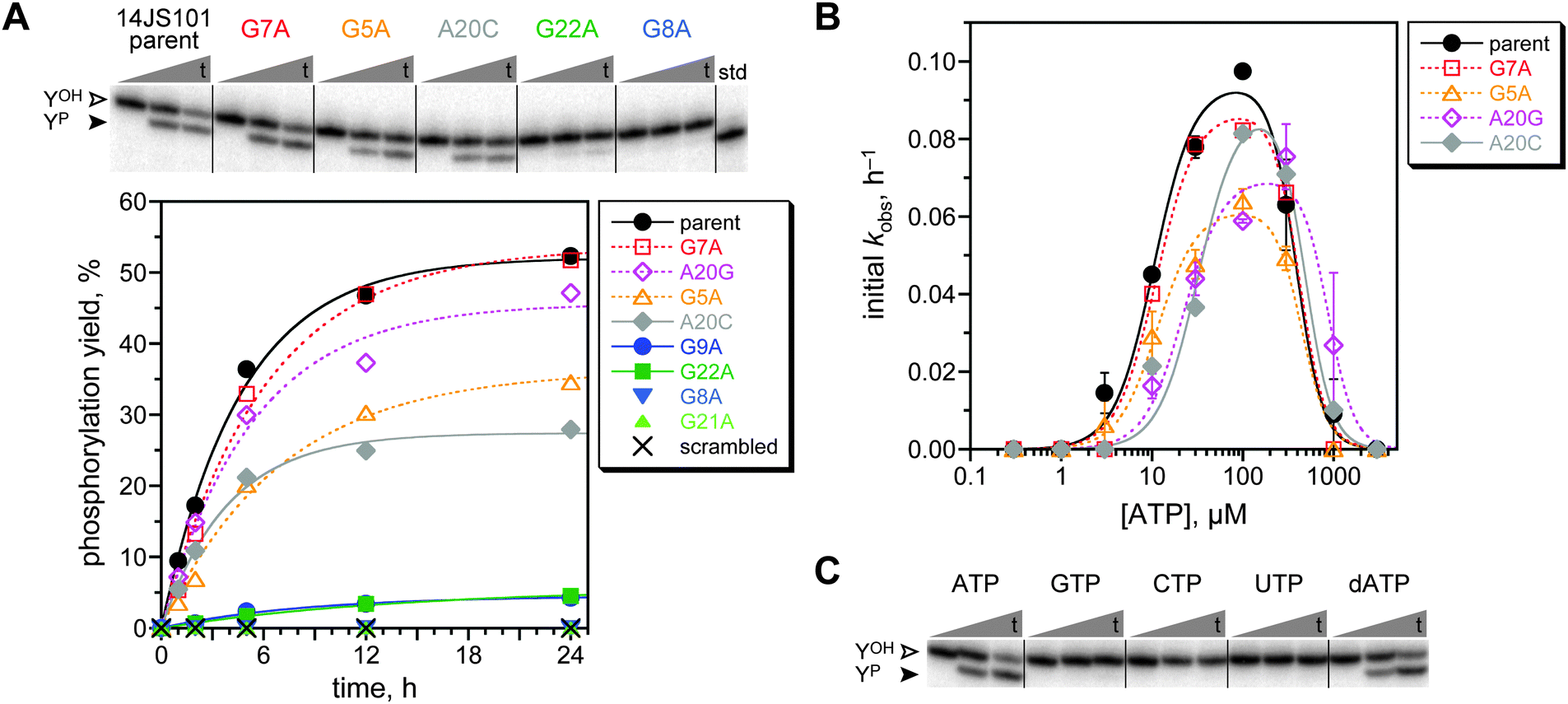 | ||
| Fig. 3 Assays of the modular 14JS101 tyrosine kinase deoxyribozyme. (A) Kinetic assays of the 14JS101 parent sequence and several mutants. For the representative PAGE image, t = 30 s, 5 h, 24 h (40 mM Tris, pH 7.5, 1 mM ZnCl2, 10 mM MnCl2, 40 mM MgCl2, 150 mM NaCl, and 30 μM ATP at 37 °C). In the plot, open symbols denote mutants expected to retain activity (innocuous mutations), and filled symbols denote mutants expected to lose activity (deleterious mutants). See text regarding A20C. Parent kobs 0.21 h−1 (first-order fit to full progress curve). (B) Assessment of Km(ATP) for the parent 14JS101 sequence and four mutants using initial-rate kinetics (0–2 h). Error bars (half of range) are shown for parent, G5A, and A20G (each n = 2); some of the error bars are smaller than the sizes of the data points. For G7A and A20C, n = 1. Curve fits used the previously described equation11 with Hill coefficients of 2 for activation and 3 for inhibition. The fit Km(ATP) values are (top to bottom in legend, μM) 10.5 ± 1.2, 11.1 ± 1.2, 11.4 ± 1.6, 22.2 ± 3.6, and 32.0 ± 5.3. (C) Assays of parent 14JS101 with all four standard NTPs and dATP. | ||
By mutational analysis, we assessed whether the ATP aptamer domain of 14JS101 contributes functionally to its catalysis. The study in which the ATP aptamer domain was initially identified also evaluated the importance of many individual nucleotides to the ATP binding.6 From those data, we chose five mutations anticipated to be deleterious for ATP binding and three mutations that should be innocuous, if the aptamer domain of 14JS101 contributes to catalysis by binding ATP. Each of these eight mutations was made separately in 14JS101, and the kinase activity of each mutant was determined (Fig. 3A). An additional mutant of 14JS101 in which the entire 27-nucleotide aptamer domain sequence was scrambled was also examined. For all but one mutant, the activity (or lack thereof) supported the conclusion that the ATP aptamer domain contributes directly to 14JS101 catalysis. For the A20C mutant, no activity was anticipated on the basis of the prior report,6 yet we observed substantial 14JS101 catalysis. Noting that the A20G mutation of the same adenosine nucleotide is both expected6 and (here) observed to allow substantial activity, we surmise that the A20C mutation is simply not as deleterious in the 14JS101 deoxyribozyme context as in the isolated ATP aptamer. Replacing the ATP aptamer module of 14JS101 with the variant MB-1 aptamer that binds only a single molecule of ATP (versus two ATP molecules for the parent aptamer)13 resulted in no 14JS101 activity under the same assay conditions (<0.5%; data not shown).
The Km for ATP of 14JS101 was determined by initial-rate kinetics to be ca. 10 μM (Fig. 3B). This value is in accord with the 6 μM Kd value for the isolated ATP aptamer,6 consistent with direct contribution of the aptamer domain to 14JS101 catalysis. The Km(ATP) was also determined for each of the active mutants. In each case, the Km value was 11–32 μM, suggesting at most a minor perturbation in the ability to bind ATP. Importantly, when 14JS101 was tested with any of GTP, CTP, or UTP, no activity was observed, whereas dATP could successfully replace ATP (Fig. 3C). These findings are also consistent with direct involvement of the ATP aptamer domain, which binds only ATP/dATP.6
From these findings, we conclude that 14JS101 is a truly modular tyrosine kinase deoxyribozyme, in which the ATP aptamer domain binds the small-molecule ATP substrate while the separate, initially random (N40) region is responsible for catalysis. An early ribozyme selection experiment attempted to include a predefined ATP aptamer domain to achieve polynucleotide kinase activity. However, the resulting aptamer domain became mutated during the selection process, and most of the corresponding ribozymes were unlikely to make functional use of the aptamer domain.10a,b Similarly, selection for a self-alkylating ribozyme using a biotinylated small-molecule substrate and a predefined biotin aptamer domain led to ribozymes that do not appear to use the (mutated) aptamer.14 In contrast to these older results, the present work establishes unequivocally that nucleic acid enzymes can use discrete aptamer and catalyst domains that together enable catalytic function. The mechanistic basis for the collaborative operation of the aptamer and catalyst domains cannot be determined from the present data. Such insights likely require high-resolution structure information, which is currently unavailable for any deoxyribozyme.15 The ATP aptamer used to identify 14JS101 binds two molecules of ATP.6,7 The available data do not establish conclusively whether or not 14JS101 requires binding of two ATP molecules. We speculate that if 14JS101 binds two ATP molecules, then only one of these molecules is properly oriented to serve as the phosphoryl donor.
A parallel goal of the present work was to evaluate whether including a predefined aptamer module leads to more robust identification of DNA catalysts. Although these experiments did successfully lead to 14JS101 as a single, explicitly modular kinase deoxyribozyme, a broader range of modular DNA catalysts was not found. Indeed, we found no deoxyribozymes at all from the analogous N30 and N50 modular selections, whereas our previous efforts with N30 and N50 random regions (but no aptamer domain) did provide DNA catalysts.11 Moreover, the kobs of 0.2 h−1 for 14JS101 is the same as the kobs of 0.2 h−1 for the fastest NTP-dependent deoxyribozyme in our previous report.11 Therefore, including a predefined aptamer module, while successful specifically for finding the 14JS101 deoxyribozyme, does not appear to be an especially helpful principle on which to design DNA catalyst selection strategies, at least for the kinase activity examined here. This conclusion is meaningful in the context of ongoing efforts to identify new deoxyribozymes that function with a wide range of small-molecule substrates. On the basis of experiments such as those reported here, we are not focusing our current efforts on identifying aptamers for small-molecule substrates followed by integrating these aptamers into DNA catalyst selections. Instead, we are primarily using random DNA regions that are tasked with simultaneously performing binding and catalysis, an approach that has been successful for a growing range of catalytic activities.1a–d However, our findings do establish experimentally that explicitly modular deoxyribozymes are possible, and for some purposes, pursuing such modularity may be advantageous.
This research was supported by grants to S.K.S. from the National Institutes of Health (R01GM065966), the Defense Threat Reduction Agency (HDTRA1-09-1-0011), and the National Science Foundation (CHE0842534). V.D. was partially supported by NIH T32GM070421. Mass spectrometry was performed on a Bruker UltrafleXtreme MALDI-TOF spectrometer purchased with NIH support (S10RR027109A).
Notes and references
- (a) A. Peracchi, ChemBioChem, 2005, 6, 1316–1322 CrossRef CAS PubMed; (b) S. K. Silverman, Chem. Commun., 2008, 3467–3485 RSC; (c) K. Schlosser and Y. Li, Chem. Biol., 2009, 16, 311–322 CrossRef CAS PubMed; (d) S. K. Silverman, Angew. Chem., Int. Ed., 2010, 49, 7180–7201 CrossRef CAS PubMed; (e) X. B. Zhang, R. M. Kong and Y. Lu, Annu. Rev. Anal. Chem., 2011, 4, 105–128 CrossRef CAS PubMed.
- (a) D. S. Wilson and J. W. Szostak, Annu. Rev. Biochem., 1999, 68, 611–647 CrossRef CAS PubMed; (b) G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791–836 CrossRef CAS PubMed; (c) G. F. Joyce, Angew. Chem., Int. Ed., 2007, 46, 6420–6436 CrossRef CAS PubMed.
- (a) R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS; (b) S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS; (c) S. K. Silverman, Nucleic Acids Res., 2005, 33, 6151–6163 CrossRef CAS PubMed; (d) K. Schlosser and Y. Li, ChemBioChem, 2010, 11, 866–879 CrossRef CAS PubMed.
- C. Khosla and P. B. Harbury, Nature, 2001, 409, 247–252 CrossRef CAS PubMed.
- F. L. Murphy and T. R. Cech, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9218–9222 CrossRef CAS.
- D. E. Huizenga and J. W. Szostak, Biochemistry, 1995, 34, 656–665 CrossRef CAS.
- C. H. Lin and D. J. Patel, Chem. Biol., 1997, 4, 817–832 CrossRef CAS.
- (a) S. D. Jhaveri, R. Kirby, R. Conrad, E. J. Maglott, M. Bowser, R. T. Kennedy, G. Glick and A. D. Ellington, J. Am. Chem. Soc., 2000, 122, 2469–2473 CrossRef CAS; (b) M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS; (c) M. N. Stojanovic and D. M. Kolpashchikov, J. Am. Chem. Soc., 2004, 126, 9266–9270 CrossRef CAS PubMed; (d) R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed; (e) E. J. Cho, L. Yang, M. Levy and A. D. Ellington, J. Am. Chem. Soc., 2005, 127, 2022–2023 CrossRef CAS PubMed; (f) J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 90–94 CrossRef CAS PubMed; (g) X. Zuo, S. Song, J. Zhang, D. Pan, L. Wang and C. Fan, J. Am. Chem. Soc., 2007, 129, 1042–1043 CrossRef CAS PubMed; (h) D. Li, B. Shlyahovsky, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2007, 129, 5804–5805 CrossRef CAS PubMed; (i) M. M. Ali and Y. Li, Angew. Chem., Int. Ed., 2009, 48, 3512–3515 CrossRef CAS PubMed; (j) X. Zuo, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2009, 131, 6944–6945 CrossRef CAS PubMed; (k) E. J. Cho, J. W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS PubMed; (l) Y. Wang, Z. Li, D. Hu, C. T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed; (m) B. Gulbakan, E. Yasun, M. I. Shukoor, Z. Zhu, M. You, X. Tan, H. Sanchez, D. H. Powell, H. Dai and W. Tan, J. Am. Chem. Soc., 2010, 132, 17408–17410 CrossRef CAS PubMed; (n) Y. Xiang and Y. Lu, Nat. Chem., 2011, 3, 697–703 CrossRef CAS PubMed; (o) K. O. Alila and D. A. Baum, Chem. Commun., 2011, 47, 3227–3229 RSC; (p) Y. Tang, B. Ge, D. Sen and H. Z. Yu, Chem. Soc. Rev., 2014, 43, 518–529 RSC.
- (a) M. Levy and A. D. Ellington, Chem. Biol., 2002, 9, 417–426 CrossRef CAS; (b) R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed; (c) M. Wieland, A. Benz, J. Haar, K. Halder and J. S. Hartig, Chem. Commun., 2010, 46, 1866–1868 RSC; (d) I. T. Seemann, V. Singh, M. Azarkh, M. Drescher and J. S. Hartig, J. Am. Chem. Soc., 2011, 133, 4706–4709 CrossRef CAS PubMed.
- (a) J. R. Lorsch and J. W. Szostak, Nature, 1994, 371, 31–36 CrossRef CAS PubMed; (b) J. R. Lorsch and J. W. Szostak, Biochemistry, 1995, 34, 15315–15327 CrossRef CAS; (c) Y. Li and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2746–2751 CrossRef CAS; (d) W. Wang, L. P. Billen and Y. Li, Chem. Biol., 2002, 9, 507–517 CrossRef CAS; (e) D. Saran, D. G. Nickens and D. H. Burke, Biochemistry, 2005, 44, 15007–15016 CrossRef CAS PubMed; (f) E. A. Curtis and D. P. Bartel, Nat. Struct. Mol. Biol., 2005, 12, 994–1000 CrossRef CAS PubMed; (g) B. Cho and D. H. Burke, RNA, 2006, 12, 2118–2125 CrossRef CAS PubMed; (h) D. Saran, D. M. Held and D. H. Burke, Nucleic Acids Res., 2006, 34, 3201–3208 CrossRef CAS PubMed; (i) E. Biondi, D. G. Nickens, S. Warren, D. Saran and D. H. Burke, Nucleic Acids Res., 2010, 38, 6785–6795 CrossRef CAS PubMed; (j) D. H. Burke and S. S. Rhee, RNA, 2010, 16, 2349–2359 CrossRef CAS PubMed; (k) E. Biondi, A. W. Maxwell and D. H. Burke, Nucleic Acids Res., 2012, 40, 7528–7540 CrossRef CAS PubMed.
- S. M. Walsh, A. Sachdeva and S. K. Silverman, J. Am. Chem. Soc., 2013, 135, 14928–14931 CrossRef CAS PubMed.
- A. Sachdeva, M. Chandra, J. Chandrasekar and S. K. Silverman, ChemBioChem, 2012, 13, 654–657 CrossRef CAS PubMed.
- M. Barbu and M. N. Stojanovic, ChemBioChem, 2012, 13, 658–660 CrossRef CAS PubMed.
- C. Wilson and J. W. Szostak, Nature, 1995, 374, 777–782 CrossRef CAS PubMed.
- J. Nowakowski, P. J. Shim, G. S. Prasad, C. D. Stout and G. F. Joyce, Nat. Struct. Biol., 1999, 6, 151–156 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparative details, in vitro selection procedure, selection progression, metal ion dependence, and mass spectrometry data. See DOI: 10.1039/c4cc04253k |
‡ The deoxyribozyme was named as 14JS101 because 14 is the round number, JS1 is the systematic alphanumeric designation for the particular selection, and 01 is the clone number. The 14JS101 sequence was 5′-ACCTGGGGGAGTATTGCGGAGGAAGGTTGAGCCCTTGCGAGAGACATGGGTCAGGACGGACAGAGGG![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -3′, where the boldface segment is the predefined ATP aptamer, the middle segment is the initially random region (40 nt), and the underlined segment is the binding arm where the DNA anchor binds. -3′, where the boldface segment is the predefined ATP aptamer, the middle segment is the initially random region (40 nt), and the underlined segment is the binding arm where the DNA anchor binds. |
| This journal is © The Royal Society of Chemistry 2014 |