 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Allylic sp3 C–H borylation of alkenes via allyl-Pd intermediates: an efficient route to allylboronates†
Hong-Ping
Deng
a,
Lars
Eriksson
b and
Kálmán J.
Szabó
*a
aDepartment of Organic Chemistry, Stockholm University, Sweden
bDepartment of Inorganic and Structural Chemistry, Stockholm University, Sweden. E-mail: kalman@organ.su.se; Web: http://www.organ.su.se/ks/ Fax: +46-8-15 49 08
First published on 26th June 2014
Abstract
Palladium catalyzed allylic C–H functionalization was performed using exocyclic alkene substrates. Multi-component synthesis of stereodefined homoallylic alcohols could be performed using a reaction sequence involving allylic C–H borylation and allylation of aldehydes.
Catalytic C–H borylation has become a practically useful synthetic method for preparation of organoboronates.1 The main reason is that these transition metal catalyzed C–H functionalization reactions can be performed under relatively mild conditions with remarkably high selectivity1b,c usually using B2Pin2 as a boronate source. The largest efforts have been focused on sp2 C–H borylation of aromatic and alkene substrates to obtain aryl/heteroaryl2 and vinyl3 boronates. However, in the last couple of years increased attention has been focused on development of sp3 C–H functionalization methods.4 These studies involved functionalization of aliphatic C–H bonds4d–h,l,m,o–q usually directed by heteroatoms, benzylic C–H bonds4i–k and there are a few examples of allylic C–H borylation4a–c as well. A selective allylic C–H borylation4a–c is particularly challenging to achieve due to two main reasons: (i) under catalytic conditions allylboronates very easily rearrange to the more stable vinylboronates.3b,c,f,4c Thus, even if the kinetic product is an allylboronate, the thermodynamic (final) product of the C–H borylation of alkenes is vinylboronate; i.e. an overall sp2 instead of sp3 C–H bond functionalization; and (ii) for non-symmetrical organometallic intermediates (e.g. allyl or alkyl-metal species) the regioselectivity of the borylation is difficult to control. Therefore, only a very few transition metal catalyzed methods are available for allylic C–H borylation of alkenes4a–c and because of (i) and (ii) the substrate scope is also very narrow.
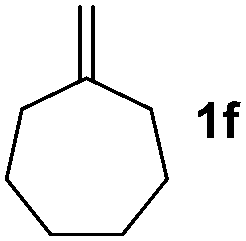
The previously developed procedures providing predominantly allylboronate products are based on C–H functionalization of simple cycloalkenes (Fig. 1). Sabo-Etienne and Caballero4a have shown that cycloheptene undergoes hydroboration and allylic C–H borylation in the presence of catalytic amounts of bis(dihydrogen)Ru complex. We have shown4b,c that simple cycloalkenes can be reacted with B2pin2 in the presence of Ir-catalysts to give allyl-Bpin compounds.
 | ||
| Fig. 1 Overview of catalytic allylic C–H borylations. | ||
Interestingly, other catalytic conditions with the above endo-cyclic alkene substrates using rhodium3g or palladium3c,f catalysts also give allyl-Bpin products in varying amounts. However, for substrates with an exo-cyclic double bond allylic C–H borylation has never been reported. This can probably be explained by the mechanistic features of the currently available Ru, Rh, Ir and Pd catalyzed methods. In all cases initial formation of an M–Bpin complex can be postulated (eqn (1)), which undergoes a syn insertion into the double bond followed by a syn selective β-hydride elimination. However, acyclic compounds can undergo unhindered rotation of the σ-bonds, and therefore the β-hydride elimination may easily result in the thermodynamically more stable vinyl–Bpin form.4c
 | (1) |
Therefore, we decided to develop a new sp3 allylic C–H borylation reaction based on an alternative mechanistic concept. We hypothesized that a Pd-catalyzed process based on initial formation of an allyl-Pd complex followed by transmetallation5 with B2pin2 may avoid the termination of the reaction with β-hydride elimination. The realization of this idea is very challenging, as closing the catalytic cycle (see bellow) requires use of oxidants, while B2pin2 is a reductant and allylboronates are sensitive to oxidation.6
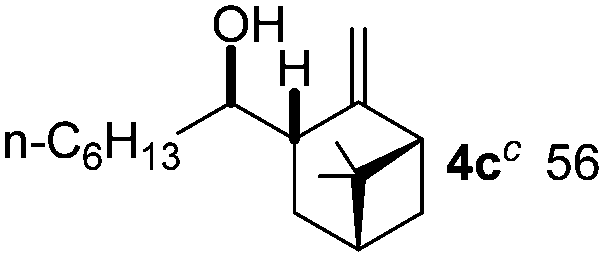
We directed the initial studies to C–H functionalization of β-pinene 1a, as these compounds readily form7 (η3-allyl)palladium complexes with stoichiometric amounts of Pd-salts. Indeed, when 1a, 3a, an appropriate oxidant and catalytic amounts of Pd(TFA)2 were mixed in (CD3)2CO, formation of borylated β-pinene was observed (Fig. 1c). Using deuterated acetone enabled us to follow the reaction by 1H NMR. The 1H NMR of the reaction mixture showed that the reaction was not completed, probably because of product inhibition. When the allylboronate product was quenched with nitro-benzaldehyde (2), the corresponding homoallylic alcohol 4a was formed selectively as a single, regio-stereoisomer.
Gratifyingly, the entire procedure with 1a, 2, 3a, the oxidant (BQ), TFA and the Pd-catalyst could be performed as a multi-component8 (or genuine one-pot) reaction (Table 1, entry 1). As we used optically active β-pinene, the multicomponent C–H borylation–allylation sequence gave an enantiomerically pure product (4a). The structure of 4a was assigned on the basis of single crystal X-ray diffraction. Subsequently, we studied the synthetic scope of the reaction. We have found that cyclic substrates with an exocyclic double bond give synthetically useful yields in the C–H borylation based allylation of aldehydes (Table 1). In most cases (except 1a) we obtained complex mixtures and low yields, when we used BQ as an oxidant. However, 2,6-dimethyl BQ (DMBQ) successfully replaced BQ. Deuterated acetone proved to be an ideal solvent in most cases as it allowed us to study the crude mixtures by 1H NMR. In some cases, the process was slow in acetone (e.g. entries 4–6 and 10) and therefore the solvent was changed to trifluoro toluene, which gave a higher reaction rate.
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | Temp. (°C)/solvent | Yieldb (%) | B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L :L |
|
a Unless otherwise stated the reactions were carried out with 1 (0.1 mmol), 2, nitro-benzaldehyde (0.2 mmol), 3a (0.2 mmol), Pd(TFA)2 (0.01 mmol), DMBQ (0.2 mmol) and TFA (0.05 mmol) in solvent (0.2–0.5 mL) for 24 h.
b Isolated yields for the linear (L) and branched (B) products together. Unless otherwise stated the branched product was isolated as a single diastereomer.
c PhCHO (entry 2) and n-C6H13CHO (entry 3) were used instead of nitro-benzaldehyde.
d d.r. = 9:1.
e Tetramethyl-benzoquinone instead of DMBQ.
f Reaction was carried out with 1h (0.2 mmol) and 2 (0.1 mmol).
g d.r. = 3:2. Ar = 4-NO2C6H4, DMBQ = 2,6-dimethylbenzoquinone.
|
||||
| 1 |

|
rt/(CD3)2CO |

|
>50:1 |
| 2 | 1a | rt/(CD3)2CO |

|
>26:1 |
| 3 | 1a | rt/(CD3)2CO |
|
>50:1 |
| 4 |
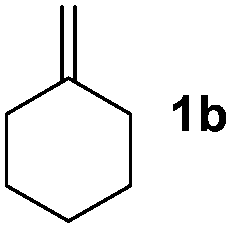
|
40/PhCF3 |

|
>50:1 |
| 5 |

|
40/PhCF3 |

|
>50:1 |
| 6 |

|
40/PhCF3 |

|
>50d:1 |
| 7e |

|
rt/(CD3)2CO |
|
>50:1 |
| 8 |
|
40/(CD3)2CO |
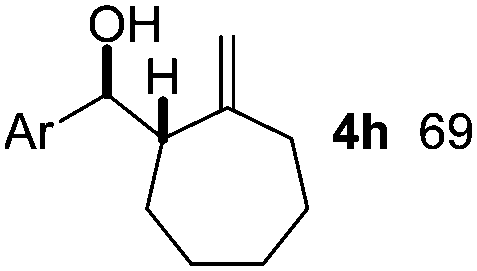
|
8:1 |
| 9 |

|
40/(CD3)2CO |

|
2.5:1 |
| 10f |

|
40/PhCF3 |
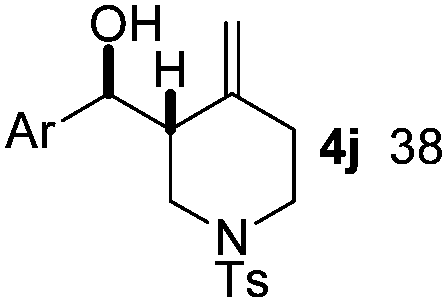
|
>50:1 |
| 11 |

|
40/(CD3)2CO |

|
15g:1 |

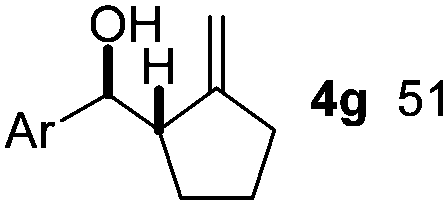
Nitro-benzaldehyde 2 could be replaced by benzaldehyde or aliphatic aldehyde (entries 2 and 3). The multicomponent reaction is still very selective but the yield was dropped (cf. entries 1–3). The reaction with six-membered ring based substrates 1b–d gave exclusively the branched allylic products 4d–e (entries 4 and 5). There are three stereogenic carbons in product 4f, thus statistically four diastereomers could be formed. However, the reaction proceeds with a remarkably high stereoselectivity, as only two diastereomers were obtained in a ratio of 9:1 (entry 6). The reactions for the five membered ring based substrate 1e proceeded faster than for the six membered ring analogs, and therefore the reactions could be conducted at rt. The best yield and selectivity were obtained, when DMBQ was replaced by tetramethyl BQ as an oxidant (entry 7). The seven membered ring based substrate 1f reacted with high yield (entry 8) but the regioselectivity was also lower than for the six-membered ring based substrates. In the case of 1g containing an eight-membered ring the regioselectivity drops to 2.5:1 (entry 9). We had a limited success with borylation of heterocyclic substrates, such as 1h (entry 10). This compound can also be transformed into 4j with high selectivity but the yield is poor and we could not improve it by extensive optimization. For acyclic analogs the yield and the selectivity drop dramatically (entry 11). For example 1i reacts very slowly and with low conversion probably because of inhibition of the Pd-catalyst. Interestingly, the reaction can be performed with diboronic acid (3b) instead of B2pin2 with a slight alteration of the reaction conditions (eqn (2)). This is a remarkable result, as it shows that highly oxidation sensitive allylboronic acids6 can also be reaction intermediates under oxidative allylic C–H borylation conditions.
 | (2) |
We suggest that the first step of the process is formation of allyl-palladium complex 5 by deprotonation and palladation of the allylic position of the substrate, such as 1b (Fig. 2).7 The subsequent step is transmetallation by B2pin2. It was shown5 that these reactions proceed easily, when weakly coordinating ligands are on Pd. This could explain that Pd(TFA)2 is an excellent catalyst for the process, while Pd(OAc)2 with the chelating acetate group is inefficient. Iwasawa and co-workers9 have recently shown that Pd–Bpin complexes are stable species. The reductive elimination of the Bpin group in 6 is supposed to be fast5 due to the strong trans influence of the Bpin ligand.10 It proceeds with a very high regioselectivity leading to the linear allylboronate product. This high regioselectivity is a prerequisite of the high selectivity of the allylation of aldehyde 2 affording the branched homoallylic product 4d. The reductive elimination involves formation of Pd(0), which has to be reoxidized at the closing of the catalytic cycle. The main role of the used quinone is reoxidation of Pd(0) to Pd(II). Added trifluoroacetic acid increases the oxidation potential of the quinones and also catalyzes the allylboration of the aldehydes.11
 | ||
| Fig. 2 Suggested catalytic cycle exemplified by substrate 1b. | ||
In summary, we have shown for the first time that allylic C–H borylation can be performed with exocyclic alkenes. Multicomponent reaction involving this new C–H borylation–allylboration sequence can be performed to obtain stereodefined homoallylic alcohols. The reaction proceeds via regioselective borylation and a subsequent regio- and stereoselective allylation. The mechanistically novel element in this reaction is that it proceeds via initial formation of an allyl-palladium intermediate, which then undergoes transmetallation with B2pin2 and a subsequent regioselective reductive elimination of the allylboronate product.
Support from the Swedish Research Council and the Knut och Alice Wallenbergs Foundation, as well as a post-doctoral fellowship for H.-P. Deng from the Wenner-Green foundation, is gratefully acknowledged. The generous gift of B2pin2 from Allychem is appreciated.
Notes and references
- (a) D. G. Hall, Boronic Acids, Wiley, Weinheim, 2011 Search PubMed; (b) I. A. I. Mkhalid, J. M. Murphy, J. H. Barnard, T. B. Marder and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; (d) T. Ishiyama and N. Miyaura, Pure Appl. Chem., 2006, 78, 1369 CrossRef CAS.
- (a) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305 CrossRef CAS PubMed; (b) I. I. B. A. Vanchura, S. M. Preshlock, P. C. Roosen, V. A. Kallepalli, R. J. Staples, J. R. E. Maleczka, D. A. Singleton and I. I. I. M. R. Smith, Chem. Commun., 2010, 46, 7724 RSC; (c) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed; (d) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS; (e) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 CrossRef CAS PubMed; (f) D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS PubMed; (g) C. C. Tzschucke, J. M. Murphy and J. F. Hartwig, Org. Lett., 2007, 9, 761 CrossRef CAS PubMed; (h) I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, R. N. Perutz and T. B. Marder, Angew. Chem., Int. Ed., 2006, 45, 489 CrossRef CAS PubMed; (i) T. Ishiyama, H. Isou, T. Kikuchi and N. Miyaura, Chem. Commun., 2010, 46, 159 RSC; (j) S. Kawamorita, H. Ohmiya and M. Sawamura, J. Org. Chem., 2010, 75, 3855 CrossRef CAS PubMed; (k) K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978 CrossRef CAS PubMed; (l) A. Ros, R. López-Rodríguez, B. Estepa, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 4573 CrossRef CAS PubMed; (m) H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 134 CrossRef CAS PubMed; (n) B. Xiao, Y.-M. Li, Z.-J. Liu, H.-Y. Yang and Y. Fu, Chem. Commun., 2012, 48, 4854 RSC; (o) Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431 CrossRef CAS PubMed.
- (a) I. A. I. Mkhalid, R. B. Coapes, S. N. Edes, D. N. Coventry, F. E. S. Souza, R. L. Thomas, J. J. Hall, S. W. Bi, Z. Y. Lin and T. B. Marder, Dalton Trans., 2008, 1055 RSC; (b) V. J. Olsson and K. J. Szabó, Org. Lett., 2008, 10, 3129 CrossRef CAS PubMed; (c) N. Selander, B. Willy and K. J. Szabó, Angew. Chem., Int. Ed., 2010, 49, 4051 CrossRef CAS PubMed; (d) T. Ohmura, Y. Takasaki, H. Furukawa and M. Suginome, Angew. Chem., Int. Ed., 2009, 48, 2372 CrossRef CAS PubMed; (e) J. Takaya, N. Kirai and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 12980 CrossRef CAS PubMed; (f) N. Kirai, S. Iguchi, T. Ito, J. Takaya and N. Iwasawa, Bull. Chem. Soc. Jpn., 2013, 86, 784 CrossRef CAS; (g) A. Kondoh and T. F. Jamison, Chem. Commun., 2010, 46, 907 RSC; (h) T. Kikuchi, J. Takagi, H. Isou, T. Ishiyama and N. Miyaura, Chem. – Asian J., 2008, 3, 2082 CrossRef CAS PubMed; (i) T. Kikuchi, J. Takagi, T. Ishiyama and N. Miyaura, Chem. Lett., 2008, 37, 664 CrossRef CAS; (j) I. Sasaki, H. Doi, T. Hashimoto, T. Kikuchi, H. Ito and T. Ishiyama, Chem. Commun., 2013, 49, 7546 RSC.
- (a) A. Caballero and S. Sabo-Etienne, Organometallics, 2007, 26, 1191 CrossRef CAS; (b) V. J. Olsson and K. J. Szabo, Angew. Chem., Int. Ed., 2007, 46, 6891 CrossRef CAS PubMed; (c) V. J. Olsson and K. J. Szabó, J. Org. Chem., 2009, 74, 7715 CrossRef CAS PubMed; (d) H. Y. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS; (e) J. M. Murphy, J. D. Lawrence, K. Kawamura, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 13684 CrossRef CAS PubMed; (f) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 12422 CrossRef CAS PubMed; (g) C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 3375 CrossRef CAS PubMed; (h) S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157 CrossRef CAS PubMed; (i) T. A. Boebel and J. F. Hartwig, Organometallics, 2008, 27, 6013 CrossRef CAS; (j) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS; (k) T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 1082 CrossRef CAS; (l) T. Ohmura, T. Torigoe and M. Suginome, J. Am. Chem. Soc., 2012, 134, 17416 CrossRef CAS PubMed; (m) T. Ohmura, T. Torigoe and M. Suginome, Organometallics, 2013, 32, 6170 CrossRef CAS; (n) T. Ohmura, T. Torigoe and M. Suginome, Chem. Commun., 2014, 50, 6333 RSC; (o) S. Kawamorita, T. Miyazaki, T. Iwai, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2012, 134, 12924 CrossRef CAS PubMed; (p) S. Kawamorita, R. Murakami, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2013, 135, 2947 CrossRef CAS PubMed; (q) T. Mita, Y. Ikeda, K. Michigami and Y. Sato, Chem. Commun., 2013, 49, 5601 RSC.
- J. M. Larsson and K. J. Szabó, J. Am. Chem. Soc., 2013, 135, 443 CrossRef CAS PubMed.
- M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 51, 13050 CrossRef CAS PubMed.
- B. M. Trost, P. E. Strege, L. Weber, T. J. Fullerton and T. J. Dietsche, J. Am. Chem. Soc., 1978, 100, 3407 CrossRef CAS.
- K. J. Szabó, in Science of Synthesis Reference Library: Multicomponent Reactions, ed. T. J. J. Müller, Thieme, Stuttgart, 2013, p. 345 Search PubMed.
- N. Kirai, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 2493 CrossRef CAS PubMed.
- J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384 CrossRef CAS PubMed.
- (a) V. Rauniyar and D. G. Hall, Angew. Chem., Int. Ed., 2006, 45, 2426 CrossRef CAS PubMed; (b) S. H. Yu, M. J. Ferguson, R. McDonald and D. G. Hall, J. Am. Chem. Soc., 2005, 127, 12808 CrossRef CAS PubMed; (c) N. Selander, S. Sebelius, C. Estay and K. J. Szabó, Eur. J. Org. Chem., 2006, 4085 CrossRef CAS; (d) N. Selander, A. Kipke, S. Sebelius and K. J. Szabó, J. Am. Chem. Soc., 2007, 129, 13723 CrossRef CAS PubMed.
Footnote |
| † CCDC 1000349. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc04151h |
| This journal is © The Royal Society of Chemistry 2014 |