 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLearning from nature: introducing an epiphyte–host relationship in the synthesis of alloy nanoparticles by co-reduction methods†
Yue
Yu
,
Qingbo
Zhang
,
Qiaofeng
Yao
,
Yi
Zhan
,
Meihua
Lu
,
Liuqing
Yang
,
Chaohe
Xu
,
Jianping
Xie
* and
Jim Yang
Lee
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 10 Kent Ridge Crescent, Singapore 119260, Singapore. E-mail: chexiej@nus.edu.sg; cheleejy@nus.edu.sg
First published on 2nd July 2014
Abstract
This communication reports an epiphytic co-reduction method which can overcome the common tendency of sequential deposition in the synthesis of alloy nanoparticles. In this method the reduction of one of the metals (the epiphyte-metal) is only turned-on and rendered more facile by the in situ generated fresh surfaces of the other metal (the host-metal).
Epiphyte is a plant which grows non-parasitically on the surface of another plant. An epiphyte derives its own resources for growth without adversely affecting the host. The relationship between the epiphyte and the host is specific and special. The epiphyte–host concept in botany is being applied to material synthesis here to overcome the problem of sequential deposition of component metals in the preparation of alloy nanoparticles (NPs). The basic premise is that certain reactions for growth of materials can be thermodynamically or kinetically promoted by the presence of a foreign surface.1 Such surface sensitivity resembles epiphytic growth and an analogous epiphyte–host relationship between two metals may be developed to support the simultaneous co-reduction of the metal precursors to homogenous alloy NPs.
The interest in alloy NPs is driven by the prospect of increased diversity and versatility of metal nanostructures through the integration of different metal NP properties or the synergy of specific component interactions.1b,2 Alloy NPs are generally prepared from the co-reduction of metal precursors. Composition uniformity is difficult in co-reduction due to the prevalence of sequential reduction.1b,2a,3 Sequential reduction is driven thermodynamically by the different electrode potentials of metals and causes the formation of core–shell NPs or other types of hetero-nanostructures.2a Sequential reduction is commonly addressed by decreasing the electrode potential difference between the component metals (e.g. through precursor complexation and/or reductant selection). This strategy may introduce complications in NP morphology control since the reduction kinetics, which affects the shape evolution of NPs,2a,3a,4 may also be altered. The epiphytic co-reduction method described in the following can be an alternative in such circumstances. Instead of narrowing the electrode potential difference, the epiphytic co-reduction method uses surface-dependent growth to control the deposition process. Specifically the reduction of one of the metals requires the presence of the surface of the other metal. The former can therefore be regarded as the epiphyte-metal, and the metal enabling the reduction of the epiphyte-metal as the host-metal. In the presence of the host-metal, the reduction of the epiphyte-metal is rendered thermodynamically more favorable than the reduction of the host-metal. With such an epiphyte–host relationship, macroscopic sequential deposition of the metals can be largely avoided because the (facile) reduction of the epiphyte-metal is regulated by the free host-metal surfaces generated in situ by the reaction system, thereby promoting the synchrony of the deposition of the two metals.
Central to the epiphytic co-reduction method is the design of a chemical reaction system which supports an epiphyte–host relationship. Underpotential deposition (UPD) provides a good implementation platform.5 UPD refers to the phenomenon where the deposition of a metal is more facile on a specific foreign metal surface than on its own; i.e. deposition occurs at a potential less negative than that on the same surface.6 UPD has been used in the synthesis of alloy surfaces5,7 or NPs8 by increasing the electrode potential of the less reducible metal. The use of UPD to establish an epiphyte–host relationship, a unique feature of this work, is conceptually different – the reduction of the UPD-metal is rendered thermodynamically more favorable by specific foreign metal surfaces. In this relationship the UPD-metal is the epiphyte-metal and the UPD-promoting foreign metal is the host-metal. As a proof of concept, Ag was selected as the epiphyte-metal since it can be deposited on many metal surfaces by UPD.6,9 Pd or Pt was selected as the host-metal in this study in view of the technological importance of Pt-group metals (PGM) and their alloys in catalysis.2a,10 The co-reduction of Ag and the PGM was carried out in the CTAB (cetyl trimethylammonium bromide, the capping agent)–AA (ascorbic acid, the reducing agent) reaction system. Cyclic voltammetry was used to determine the reduction potential of Ag(I) in this reaction system (Fig. S1, ESI†). The electrode potential measured as such was −0.12 V vs. the Ag|AgCl reference electrode (or 0.385 V vs. SHE). The value is more negative than the typical electrode potential of Ag+/Ag (0.7996 V) as a result of the formation of dissolved silver halide species with CTAB.11 The reducing agent AA was inadequate to reduce the Ag(I) in our reaction system at room temperature.12 In the presence of a Pt or a Pd surface, however, Ag(I) can be reduced at higher potentials, with UPD shifts (ΔEUPD) of 0.336–0.435 V on Pt6,9,13 and 0.296 V on Pd9 respectively. Ag(I) reduction was not only turned on by the presence of Pt or Pd host-metal surfaces but was also more favorable compared with the host-metal reduction (standard electrode potentials of PtBr62−/Pt and PdBr42−/Pd are 0.613 V and 0.49 V respectively). An epiphyte–host relationship was therefore established. In this reaction system Ag(I) could not be reduced by AA in the presence of CTAB at room temperature, unless a Pd or a Pt host-metal surface was available to promote Ag UPD.
The proposed mechanism for alloy NP formation by the epiphytic co-reduction approach is illustrated in Scheme 1A. Upon mixing of the metal precursors with the reducing agent, the host-metal precursor was reduced first and the nuclei of the host-metal were formed (Scheme 1A–I). Once a fresh host-metal surface was formed, the reduction of the epiphyte-metal was turned on to facilely deposit the epiphyte-metal on the host-metal surface (Scheme 1A–II). All the while the reduction of the host-metal continued (albeit less easily than the UPD of Ag(I)) and host-metal ad-atoms or clusters were deposited on the growing NPs. New host-metal surfaces were generated, initiating another round of epiphytic metal reduction and deposition (Scheme 1A–III and A–IV), and the cycle was repeated. In this way the two component metals were reduced in tandem to form NPs with a uniform composition throughout (Scheme 1A–V).
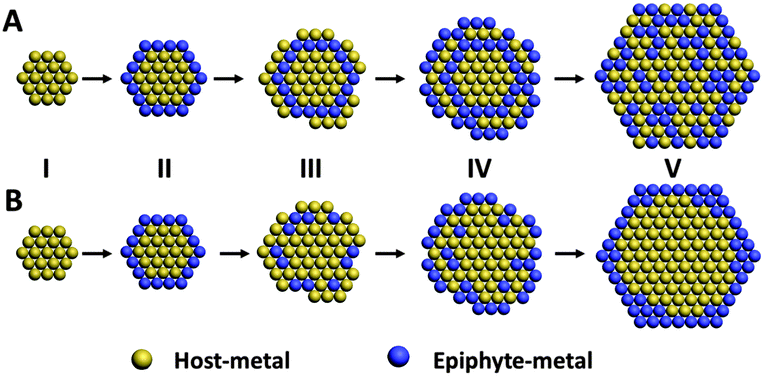 | ||
| Scheme 1 (A) Schematic illustration of the formation of alloy NPs by the epiphytic co-reduction method. (I) Reduction of the host-metal precursor to nucleate a host-metal surface. (II) The reduction of the epiphyte-metal on the freshly formed host metal surface, depositing the epiphyte-metal onto the latter. (III)–(IV) The reduction and deposition of host-metal continued, generating fresh surfaces for the reduction and deposition of the epiphyte-metal. (V) Repeats of steps (III) and (IV) lead to the formation of alloy NPs. (B) Formation of core–shell NPs due to the dealloying of alloy NPs if there is an aggressive galvanic replacement reaction between the epiphyte metal and the host metal precursor. The more reactive metal component (the epiphyte-metal in this illustration) is oxidized and dissolved away during the formation of alloy NPs resulting in its very low content in the composite NPs. | ||
Composition analysis revealed that the NPs were solid solutions of the component metals (alloys). Elemental mapping showed uniform distributions of Ag and Pt (or Pd) throughout the bulk of the NPs (Fig. 1A and B). Cross-sectional line scans also produced superimposable signals of Ag and Pt (or Pd) indicating the homogeneity of alloy composition. XRD further corroborated the formation of alloy NPs since the Bragg angles of the diffraction peaks are immediate between those of fcc-Ag and fcc-Pt (or fcc-Pd); without the presence of monometallic Ag or Pt (or Pd) (Fig. 1A-4 and 1B-4).
 | ||
| Fig. 1 Compositional analyses of (A) Ag–Pt alloy NPs and (B) Ag–Pd alloy NPs with 50 atom% of Ag. Sets 1–4 are STEM images, STEM-EDX line-scans, elemental mapping images and XRD patterns. | ||
The composition of the alloy NPs could be varied by changing the metal precursor concentrations in the reaction mixture. The ability to adjust alloy composition is important since many alloy properties are composition-dependent. The XRD patterns of Ag–Pt and Ag–Pd alloy NPs with different compositions are shown in Fig. 2A and B. With the increase of the Ag atom% in the precursor mixture, the diffraction peaks of alloy NPs shifted progressively from the Bragg angles of Pt (or Pd) to those of Ag; as expected of Vegard's law. Quantification of the bulk (by EDX) and surface (by XPS) compositions of Ag–Pt and Ag–Pd NPs (Table S2, ESI†) showed good congruence of the measured values; yet another evidence for the homogeneity of composition and the absence of heterostructures. A plot of alloy Ag atom% versus the Ag content of the growth solution (Fig. 2C and D) was linear until certain threshold values were exceeded (∼55 atom% for Ag–Pt NPs and ∼35 atom% for Ag–Pd NPs for 50% and 60% Ag atom% in the precursor mixture respectively). Thereafter the alloy composition started to level off. The leveling of alloy composition is an indication that the host-metal surface has reached its capacity limit for Ag(I) reduction. Since Ag(I) reduction in the current reaction system was based on UPD on in situ generated host-metal surfaces, the capacity limit should correspond to the maximum coverage of the epiphyte-metal on the host-metal surface, which is usually a monolayer.6 New host-metal surfaces have to be generated to sustain the epiphytic reduction of Ag(I) by UPD after reaching monolayer coverage on the host-metal. Below the saturation limit the alloy Ag content could be increased in proportion to the Ag content in the precursor mixture.
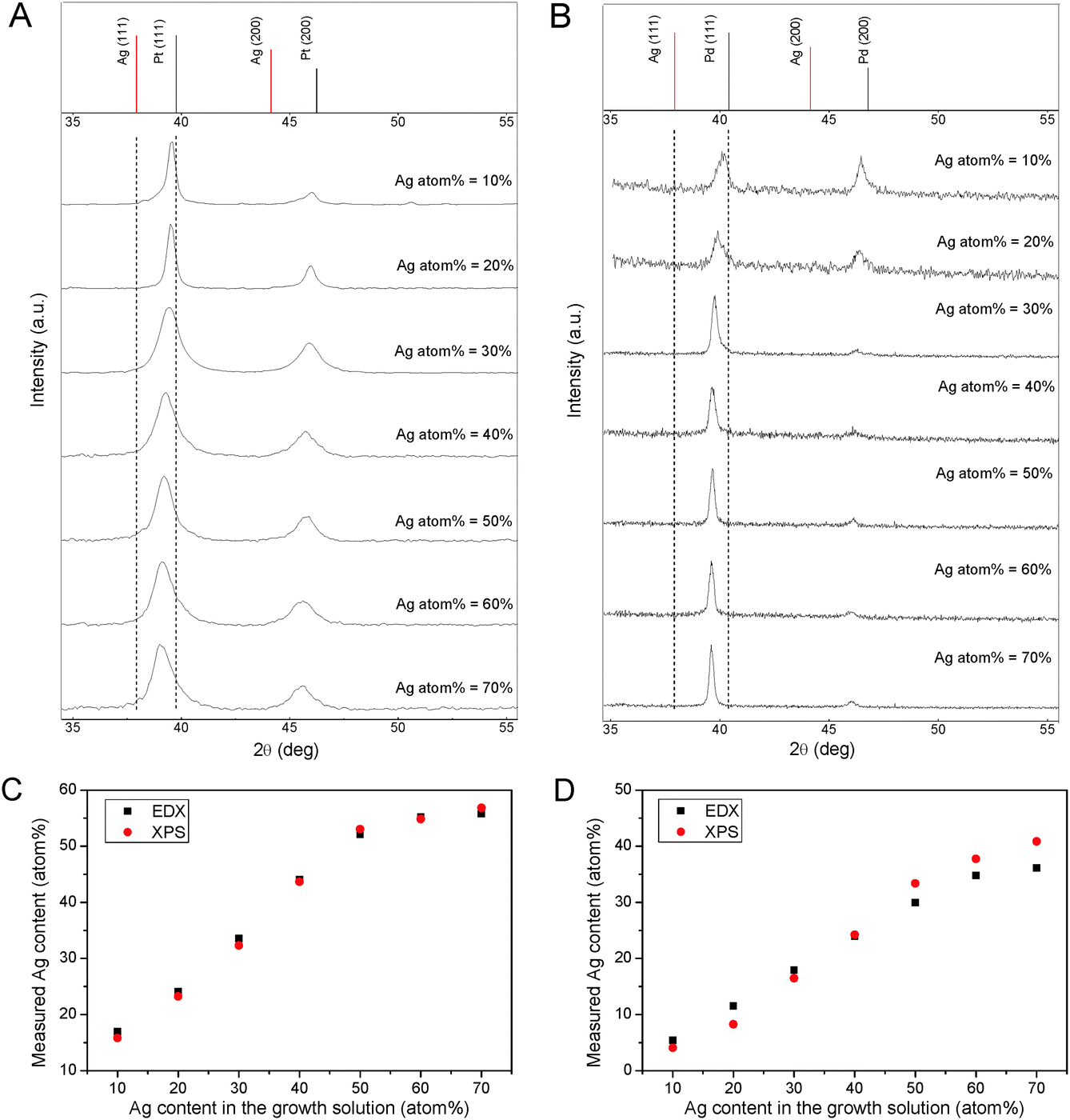 | ||
| Fig. 2 Tuning the composition of the alloy NPs. XRD patterns of (A) Ag–Pt and (B) Ag–Pd alloy NPs in different compositions (with reference patterns from the following JCPDS database: Ag, 00-001-1164; Pd, 00-001-1201; and Pt, 00-004-0802). Plots of EDX and XPS measurements of Ag atom% in (C) Ag–Pt NPs and (D) Ag–Pd NPs versus the Ag content in the growth solution (the experimentally measured values are also shown in Table S2, ESI†). | ||
The Ag content in the alloy NPs was close to the Ag content in the precursor mixture for the Ag–Pt NPs, but lower for the Ag–Pd NPs. The lower Ag content in the Ag–Pd NPs could be attributed to a surreptitious dealloying process where the more reactive component of the alloy NPs (Ag in this case) was galvanically oxidized by the precursor of the less reactive metal component (Pt or Pd in this case) due to the higher electrode potentials of Pt and Pd relative to Ag (Scheme 1B).14 The prevalence of dealloying was revealed in an experiment when a solution of the precursor of the less reactive metal (Pt or Pd) was added to a solution of washed alloy NPs. The latter was prepared by redispersing the as-synthesized alloy NPs (after centrifugation to remove AA) in aqueous CTAB solution. The Ag atom% in both Ag–Pt and Ag–Pd alloy NPs decreased after treatment with the Pt(IV) or the Pd(II) precursor solution (Table S3, ESI†). The % decrease in Ag content was higher for the Ag–Pd alloy NPs than the Ag–Pt alloy NPs. Since the external reducing agent (AA) has been completely removed in the experiment, the decrease in the Ag atom% could only be caused by the galvanic oxidation of Ag0 in the alloy by Pt(IV) or Pd(II) precursor ions. The greater decrease in the Ag atom% for the Ag–Pd NPs indicates that Ag0 in Ag–Pd NPs was more susceptive to galvanic oxidation by Pd(II) ions. A larger amount of Ag0 was therefore lost from the alloy structure by oxidative dissolution. Alloy formation by an epiphytic co-reduction approach may not even be possible if dealloying is extensive and pervasive. This is the case for the Ag–Au system where composition analysis detected the formation of Ag-coated Au NPs and not Ag–Au alloy NPs (Table S4 and Fig. S4, ESI†) despite the fact that Au is a host for Ag UPD12a,c and satisfies the epiphyte–host relationship (UPD shift, ΔEUPD, of Ag on Au is 0.505–0.55 V;9,13 the standard electrode potential of AuBr4−/Au is 0.854 V). A strong dealloying reaction by the Ag–Au galvanic replacement reaction could completely oxidize the Ag0 in Ag–Au NPs. The formation and depletion of Ag0 would continue until the entire stock of the Au precursor was exhausted by AA reduction; ending in a final Ag UPD layer. Such Ag-layer-coated-Au nanostructures are commonly found in the shape-controlled synthesis of Au NPs by Ag UPD.12
The epiphytic co-reduction approach is also able to vary the shape of the alloy NPs while keeping the alloy composition fixed. Since morphology also influences the properties of alloy NPs,2a,10,15 the ability to delink composition and morphology in an alloy NP synthesis is of great value to the determination of reliable composition–property and morphology–property relationships. NP morphology is typically manipulated through growth kinetics control in solution based syntheses.12b,16 In a conventional co-reduction synthesis, growth kinetics control inevitably leads to concurrent changes in composition and morphology. In an epiphytic co-reduction synthesis, on the other hand, the changes in reduction kinetics may change the deposition rates (with some accompanying morphology changes); but the epiphyte–host relationship is still applicable over a wide range of conditions. We have shown earlier that the alloy NP composition in an epiphyte–host growth is determined by the precursor ratio, and as such is unvarying as the shape evolves. To prove this we varied the growth kinetics by several routes, e.g., adjusting the reducing power of AA (with addition of HCl), introducing complexing counter ions (Br− in the form of NaBr), or varying the capping agent (CTAB) concentration. The resulting Ag–Pt and Ag–Pd NPs obtained at the same precursor concentration ratio did not show significant composition variations (Fig. S5 and Table S5, ESI†). The shape of the NPs, on the other hand, could be varied significantly due to the changes in growth kinetics (Fig. 3 using the Ag–Pd NPs as an example). Specifically Ag–Pd NPs were transformed from truncated octahedrons and truncated bipyramids (with exposure of both {100} and {111} facets) (Fig. 3A) to octahedrons and triangular plates ({111} facets dominated) (Fig. 3B) by the addition of HCl (16 mM). In the presence of NaBr (16 mM), the Ag–Pd NPs were cubes and right bipyramids dominated by {100} facets (Fig. 3C).
 | ||
| Fig. 3 Morphology tuning of the alloy NPs. TEM images of Ag–Pd NPs prepared under different reaction conditions: (A) the reference condition (see Table S5, ESI†), (B) with the addition of 16 mM HCl, and (C) with the addition of 16 mM NaBr. Ag–Pd NPs were prepared at a precursor Ag content of 30 atom%. Blue and yellow colors in the geometric models in (B) are {111} and {100} facets respectively. | ||
We have developed an epiphytic co-reduction approach for the synthesis of alloy NPs. Central to this approach is the design of a reaction system which can establish an epiphyte–host relationship in the co-reduction of the component metals. In this method the reduction of the epiphyte-metal is constrained by the availability of in situ formed host-metal surfaces. It is however a more facile process than the reduction of the host-metal once it occurs. The usual tendency for sequential reduction is effectively suppressed; promoting the formation of homogenous alloy NPs over heterostructures. The alloy NP composition can be varied by the precursor metal ratio in the reaction mixture up to some threshold values. Dealloying due to galvanic replacement reactions may limit the presence of the more active component in the alloy NPs and in extreme cases, inhibit the formation of alloy structures. The epiphyte co-reduction approach is also able to effectively decouple composition and morphology control in NP synthesis, allowing alloy NPs with the same composition but different morphologies to be formed.
This work was financially supported by the Ministry of Education, Singapore, under Grant R-279-000-349-112.
Notes and references
- (a) D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2010, 132, 6280 CrossRef CAS PubMed; (b) D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044 CrossRef CAS PubMed.
- (a) Z. M. Peng and H. Yang, Nano Today, 2009, 4, 143 CrossRef CAS; (b) Q. B. Zhang, J. Y. Lee, J. Yang, C. Boothroyd and J. X. Zhang, Nanotechnology, 2007, 18, 245605 CrossRef; (c) Q. B. Zhang, J. P. Xie, J. Liang and J. Y. Lee, Adv. Funct. Mater., 2009, 19, 1387 CrossRef CAS.
- (a) J. Gu, Y. W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050 RSC; (b) J. Yang and J. Y. Ying, Nat. Mater., 2009, 8, 683 CrossRef CAS PubMed.
- X. W. Liu, D. S. Wang and Y. D. Li, Nano Today, 2012, 7, 448 CrossRef CAS.
- D. Liang and G. Zangari, Langmuir, 2014, 30, 2566 CrossRef CAS PubMed.
- E. Herrero, L. J. Buller and H. D. Abruna, Chem. Rev., 2001, 101, 1897 CrossRef CAS PubMed.
- (a) J. J. Mallett, U. Bertocci, J. E. Bonevich and T. P. Moffat, J. Electrochem. Soc., 2009, 156, D531 CrossRef CAS; (b) E. Rouya, G. R. Stafford, U. Bertocci, J. J. Mallett, R. Schad, M. R. Begley, R. G. Kelly, M. L. Reed and G. Zangari, J. Electrochem. Soc., 2010, 157, D396 CrossRef CAS; (c) J. J. Mallett, W. Shao, D. Liang and G. Zangari, Electrochem. Solid-State Lett., 2009, 12, D57 CrossRef CAS.
- Y. Jiang, Y. Jia, J. Zhang, L. Zhang, H. Huang, Z. Xie and L. Zheng, Chem. – Eur. J., 2013, 19, 3119 CrossRef CAS PubMed.
- K. A. Soliman and L. A. Kibler, Electrochim. Acta, 2007, 52, 5654 CrossRef CAS.
- N. Tian, J. Xiao, Z.-Y. Zhou, H.-X. Liu, Y.-J. Deng, L. Huang, B.-B. Xu and S.-G. Sun, Faraday Discuss., 2013, 162, 77 RSC.
- X. Guo, Q. Zhang, Y. Sun, Q. Zhao and J. Yang, ACS Nano, 2012, 6, 1165 CrossRef CAS PubMed.
- (a) M. Z. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192 CrossRef CAS PubMed; (b) M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783 RSC; (c) M. L. Personick, M. R. Langille, J. Zhang and C. A. Mirkin, Nano Lett., 2011, 11, 3394 CrossRef CAS PubMed.
- D. M. Kolb, M. Przasnys and H. Gerische, J. Electroanal. Chem., 1974, 54, 25 CrossRef CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892 CrossRef CAS PubMed.
- (a) Y. Yu, Q. B. Zhang, B. Liu and J. Y. Lee, J. Am. Chem. Soc., 2010, 132, 18258 CrossRef CAS PubMed; (b) N. A. Kotov and F. Stellacci, Adv. Mater., 2008, 20, 4221 CrossRef CAS; (c) T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781 CrossRef CAS PubMed.
- (a) Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed; (b) Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153 CrossRef CAS; (c) A. R. Tao, S. Habas and P. D. Yang, Small, 2008, 4, 310 CrossRef CAS; (d) Y. Yu, Q. B. Zhang, X. M. Lu and J. Y. Lee, J. Phys. Chem. C, 2010, 114, 11119 CrossRef CAS; (e) Y. Yu, Q. Zhang, J. Xie and J. Y. Lee, Nat. Commun., 2013, 4, 1454 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, electrochemical measurements and additional compositional analyses. See DOI: 10.1039/c4cc04132a |
| This journal is © The Royal Society of Chemistry 2014 |