 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective photocatalytic oxidation of benzene for the synthesis of phenol using engineered Au–Pd alloy nanoparticles supported on titanium dioxide†
Ren
Su
a,
Lokesh
Kesavan
b,
Mads M.
Jensen
c,
Ramchandra
Tiruvalam
d,
Qian
He
d,
Nikolaos
Dimitratos
be,
Stefan
Wendt
a,
Marianne
Glasius
c,
Christopher J.
Kiely
d,
Graham J.
Hutchings
*be and
Flemming
Besenbacher
*a
aInterdisciplinary Nanoscience Centre (iNANO) and Department of Physics and Astronomy, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: fbe@inano.au.dk
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
cDepartment of Chemistry and Interdisciplinary Nanoscience Centre (iNANO), Aarhus University, Langelandsgade 140, DK-8000 Aarhus C, Denmark
dDepartment of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, 18015-3195, Bethlehem, Pennsylvania, USA
eThe UK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Oxfordshire, OX11 0FA, UK
First published on 11th July 2014
Abstract
The selectivity of photocatalytic phenol production from the direct oxidation of benzene can be enhanced by fine adjustment of the morphology and composition of Au–Pd metal nanoparticles supported on titanium dioxide thereby suppressing the decomposition of benzene and evolution of phenolic compounds.
Phenol, as an important precursor in the production of numerous resins and drugs, is produced on a large scale by the petroleum industry and the demand for phenol keeps increasing rapidly.1 Conventional synthesis of phenol involves the partial oxidation of isopropylbenzene (cumene) via the Hock rearrangement.1 Though mild reaction conditions and relatively inexpensive raw materials are required to operate the synthesis, the excessive amount of acetone produced as a by-product is a problem. Since the cumene precursor is mainly produced by the Friedel–Crafts alkylation of benzene,2 a synthesis route that directly converts benzene to phenol would be ideal to replace the indirect partial oxidation process.3,4
Whilst benzene can be easily oxidised in air at high temperature even in the absence of a catalyst, the phenol produced is further oxidised to CO2 at a much faster rate, which limits the potential conversion rate and selectivity. The utilisation of oxidants (e.g., H2O2 and N2O) in combination with heterogeneous catalysts has shown promising conversion rates and selectivity to phenol.3,4 However, the use of such expensive oxidants has limited progress in the direct oxidation process.
Heterogeneous photocatalysis has shown huge potential in organic synthesis and can provide an alternative approach for the synthesis of phenol.5,6 The OH˙ radicals can be generated on the surface of a photocatalyst under suitable irradiation conditions, suggesting that no additional oxidation reagents are required for the conversion of benzene to phenol. However, the photo-generated radical species generally show poor selectivity, which results in the complete decomposition of benzene to CO2 and several unwanted phenolic compounds (e.g., hydroquinone).7 Moreover, complete decomposition of phenolic compounds is generally faster than that of phenol, which may decrease the selectivity to phenol indirectly.8 Therefore, it is of great interest to promote the formation of phenol and “switch-off” other undesired reactions (see Scheme 1). Although the selectivity can be tuned by the addition of oxidants or tweaking the reaction conditions (i.e., solvent identity, addition of phenol),9–11 tuning the surface properties of the photocatalyst seems to be the most promising and intuitive approach to improve phenol formation.12,13 Here we show that exercising a degree of control over the co-catalyst nanoparticle (NP) morphology supported on TiO2 can significantly tune the product selectivity for photocatalytic benzene oxidation.
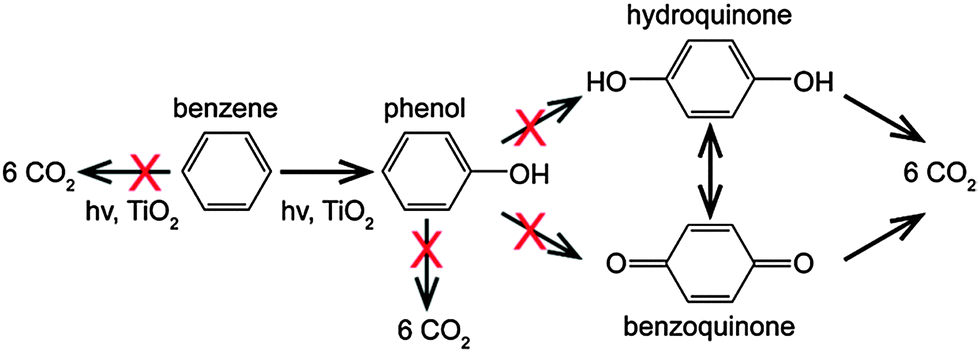 | ||
| Scheme 1 Photocatalytic oxidation pathways of benzene. The crosses indicate the unwanted reaction pathways. | ||
The co-catalysts investigated were a systematic series of metal NPs ranging from monometallic Au and Pd to random Au–Pd alloys and core–shell configurations, which were synthesised via colloidal methods and subsequently supported on a TiO2 (Degussa P25) photocatalyst by a sol-immobilisation process.14 All the metal/TiO2 samples have a nominal metal loading of 1 wt%, and some representative high angle annular dark field-scanning transmission electron microscope (HAADF-STEM) images of a sub-set of these materials are shown in Fig. 1. For the Au1Pd1 alloy (Fig. 1(b)), a homogeneous distribution of Au and Pd atoms is obvious when compared to the monometallic Au sample (Fig. 1(a)). Furthermore, the concentric layered structures of the Pdshell–Aucore and Aushell–Pdcore NPs are clearly visible in Fig. 1(c) and (d) respectively. Statistical analysis revealed that the metal NPs in all variants of the metal/TiO2 samples have a characteristic mean size of ∼4–5 nm with a narrow size distribution (Fig. S1 in ESI†).14 Furthermore, we have confirmed that the physical parameters of the TiO2 support remained unchanged after immobilisation of the metal NPs (Fig. S2 in ESI†).14
 | ||
| Fig. 1 Representative HAADF-STEM images of (a) Au, (b) Au1Pd1 random alloy, (c) Pdshell–Aucore and (d) Aushell–Pdcore NPs sol-immobilised on TiO2. The Aushell–Pdcore and Pdshell–Aucore NPs are labelled as AuSPdC and PdSAuC, respectively. | ||
We have carried out benzene oxidation in the liquid phase at room temperature (catalyst loading of 5 mg L−1) with all metal/TiO2 photocatalysts described above. A well-defined UV LED light source was used for all photocatalytic reactions (Fig. S3 in ESI†).14 The evolution of liquid phase and gas phase products was analysed by in situ UV-vis and mass spectrometry, respectively, as summarised in Fig. 2 (Fig. S4–S6 in ESI†).14 Additionally, ultra high performance liquid chromatography coupled by an electrospray ionisation inlet to a quadrupole time-of-flight mass spectrometry (UHPLC-ESI-QTOF-MS) analysis confirmed that the products were mainly phenolic compounds (phenol and hydroquinone) with short-chain carboxylic acids as minor impurities (Fig. S7 in ESI†).14 By monitoring the sextet of peaks at λ ∼ 250 nm, it was evident that benzene oxidation took place as soon as UV irradiation was commenced for all photocatalysts (Fig. 2(a)). However, the spectra varied significantly depending on the composition and morphology of the co-catalyst NPs (Fig. S5 in ESI†).14 When bare TiO2 was used for benzene oxidation, phenol (indicated by peak P, λmax = 270 nm) and hydroquinone (indicated by peak H, λmax = 289 nm) were the main product species detected in the liquid phase. In addition, some polymerisation of the phenolic compounds was observed, resulting in an increase of absorption at λ > 300 nm. It was found that hydroquinone formation and further polymerisation of phenolic compounds could be suppressed by using Au as a co-catalyst on the TiO2, but the generation of phenol was also unfortunately inhibited. Interestingly, by replacing the monometallic Au co-catalyst with Aushell–Pdcore NPs, an optimum evolution of phenol was achieved while simultaneously hindering the formation of hydroquinone. Furthermore, the polymerisation processes were found to be negligible when metal co-catalysts were employed. We also compared the complete decomposition of benzene and phenol during photo-oxidation for all samples by following the CO2 evolution, as shown in Fig. 2(b) and (c), respectively. The CO2 evolution from benzene can be strongly suppressed by the presence of metal NPs on the TiO2 surface (Fig. 2(b)), suggesting that the metal NPs not only facilitate the electron–hole separation but are also involved in the photo-oxidation reactions, where the partial oxidation of benzene was improved and the complete decomposition of benzene was inhibited.14,15 Surprisingly, the co-catalyst, irrespective of the variant used, has a negligible effect on reduction of the complete oxidation of phenol when compared to that of the pristine TiO2 (Fig. 2(c)).
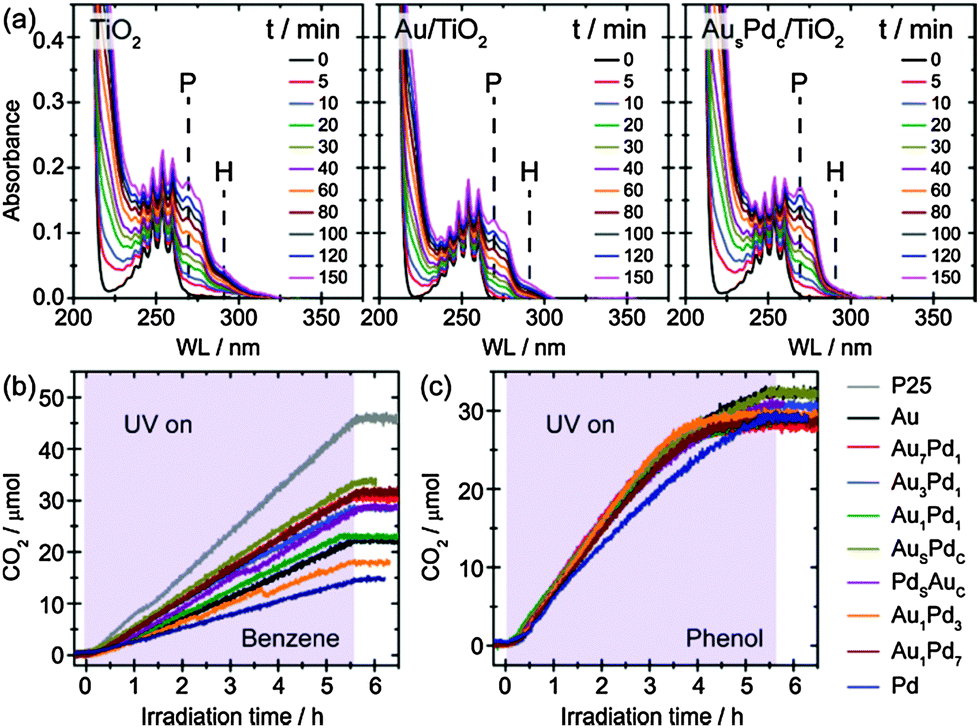 | ||
| Fig. 2 (a) Representative UV-vis spectra time sequences recorded during benzene oxidation using pristine TiO2, Au/TiO2 and Aushell–Pdcore/TiO2, respectively, as photocatalysts. P and H denote phenol and hydroquinone, respectively. (b) and (c) Photocatalytic CO2 evolution from both benzene and phenol oxidation using pristine TiO2 and various metal/TiO2 photocatalysts. The Aushell–Pdcore and Pdshell–Aucore NPs are labelled as AuSPdC and PdSAuC in all figures. | ||
Fig. 3(a) and (b), based on data extracted from UV-vis spectra, depict the effectiveness of the various co-catalyst NPs in evolving phenol and hydroquinone respectively (Fig. S6 and Table S1 in ESI†).14 Whilst monometallic Au and Pd NPs decreased the phenol evolution rates, the Au-rich and Pd-rich alloy NPs (i.e., Au7Pd1, Au3Pd1, Au1Pd3, and Au1Pd7) exhibited enhanced phenol generation rates compared to that of pure TiO2. Note however that the hydroquinone evolution rates remained similar to that of pure TiO2 for the same sub-set of photocatalysts. Surprisingly, the Au1Pd1 alloy NPs supported on TiO2 showed the worst performance in terms of generation of both phenol and hydroquinone. We also discovered that the selective oxidation of benzene depends on the nanostructure on the metal NPs. The Aushell–Pdcore NPs promoted phenol production and simultaneously decreased the formation of hydroquinone, whereas in comparison the Pdshell–Aucore NPs quenched both reactions. The inhibition of hydroquinone formation may improve the selectivity to phenol as phenolic compounds are less stable than phenol.8 We further plotted the phenol-to-hydroquinone ratio generated as a function of the co-catalyst composition/morphology (Fig. 3(c)). Noticeably, the Aushell–Pdcore NPs supported on TiO2 achieved the highest selectivity of the liquid phase products compared to the other co-catalyst formulations. Furthermore, we found that the production rate of phenol can be further improved by increasing the loading of the photocatalyst (see Fig. S8 in ESI†).14 Although the photocatalytic conversion of benzene is still relatively low (∼30%), it shows good potential for future development and use in the direct synthesis of high purity phenol.
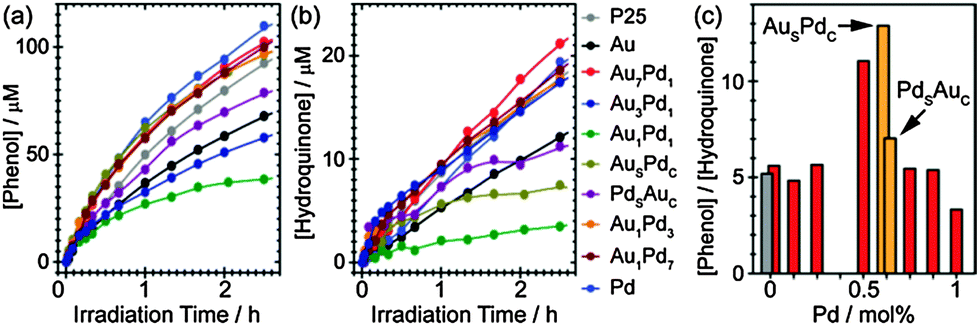 | ||
| Fig. 3 Evolution of (a) phenol and (b) hydroquinone using different metal NPs supported on TiO2 as photocatalysts. (c) The effect of co-catalyst NP identity on the selective conversion to phenol. The grey and red bars in (c) represent bare TiO2 and Au–Pd alloy supported on TiO2, respectively. The Aushell–Pdcore and Pdshell–Aucore NPs are labelled as AuSPdC and PdSAuC. | ||
In summary, an oxidative photocatalysis approach for the direct synthesis of phenol from benzene has been demonstrated. Furthermore, the need to engineer the metal co-catalyst composition and morphology has been shown to be critical for fine tuning the selectivity of this benzene oxidation reaction. The presence of co-catalysts has also been shown to have beneficial effects on inhibiting (i) the polymerisation of phenolic products and (ii) the complete oxidation of benzene. Specifically, the Aushell–Pdcore variant NPs supported on TiO2 exhibited the best photocatalytic performance by simultaneously increasing the phenol evolution rate and decreasing the hydroquinone generation rate. We anticipate that in-depth investigations on the role of metal co-catalysts will further boost the performance of photocatalysed selective oxidation reactions.
The authors acknowledge the CMC (Denmark National Research Foundation, DNRF 93), the Danish Strategic Research Council, the Carlsberg Foundation, an advanced grant from ERC (FB) and the EPSRC, and NSF for financial support. We also thank the EPSRC for funding via the UK Catalysis Hub EP/K014854/1.
Notes and references
- M. Weber, M. Weber and M. Kleine-Boymann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI: 10.1002/14356007.a19_299.pub2 Search PubMed.
- J. K. Groves, Chem. Soc. Rev., 1972, 1, 73 RSC.
- J.-H. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A.-H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793 CAS.
- G. I. Panov, CATTECH, 2000, 4, 18 CrossRef CAS.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- K. I. Shimizu, T. Kaneko, T. Fujishima, T. Kodama, H. Yoshida and Y. Kitayama, Appl. Catal., A, 2002, 225, 185 CrossRef CAS.
- X. Wang, L. Sø, R. Su, S. Wendt, P. Hald, A. Mamakhel, C. Yang, Y. Huang, B. B. Iversen and F. Besenbacher, J. Catal., 2014, 310, 100 CrossRef CAS PubMed.
- K. Zama, A. Fukuoka, Y. Sasaki, S. Inagaki, Y. Fukushima and M. Ichikawa, Catal. Lett., 2000, 66, 251 CrossRef CAS.
- H. Park and W. Choi, Catal. Today, 2005, 101, 291 CrossRef CAS PubMed.
- Y. Ide, M. Matsuoka and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 16762 Search PubMed.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, RSC Adv., 2013, 3, 5121 RSC.
- T. Marino, R. Molinari and H. Garcia, Catal. Today, 2013, 206, 40 CrossRef CAS PubMed.
- Detailed sample preparation and characterisation (HAADF-STEM, UV-vis spectra, UHPLC-ESI-QTOF-MS spectra, and the high catalyst loading test) are available in the ESI†.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2012, 6, 6284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV-vis spectra, and UHPLC-ESI-QTOF-MS data. See DOI: 10.1039/c4cc04024d |
| This journal is © The Royal Society of Chemistry 2014 |