
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic coupling mediated by furan, thiophene, selenophene and tellurophene in a homologous series of organic mixed valence compounds†
Ann Christin
Jahnke
,
Mariana
Spulber
,
Markus
Neuburger
,
Cornelia G.
Palivan
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 4th August 2014
Abstract
Charge delocalization in the mixed-valent monocationic forms of phenothiazine-decorated chalcogenophenes is explored by cyclic voltammetry, optical absorption and EPR spectroscopy. Single units of furan, thiophene, selenophene and tellurophene are found to mediate electronic coupling between the phenothiazines attached to their 2- and 5-positions roughly equally well. Electronic communication seems to occur mostly through the butadiene-like backbone of the chalcogenophenes.
The ability of π-conjugated oligomers and polymers to become highly conducting upon oxidation is of key importance for various (opto)electronic applications.1 Numerous studies have investigated charge delocalization and charge transport phenomena in oxidized oligo- and polythiophenes.2 Lately there has been increasing interest in oligomers and polymers of the other chalcogenophenes, in particular selenophene,3 but also furan and tellurophene.4 However, despite important recent progress, the synthesis of oligofurans and oligotellurophenes remains nontrivial.5 From a fundamental perspective and with the application potential of furan- or tellurophene-based materials in mind, it seemed worthwhile to explore to what extent an unpaired electron can delocalize over individual chalcogenophene units with the heteroatoms varying along the series O, S, Se, Te. Charge transport in oligomers and charge delocalization in monomers are different issues, but in order to tailor the properties of an oligomer or polymer it seems desirable to understand the electronic structure of its monomeric building blocks as detailed as possible.
Mixed valence compounds have been frequently employed for exploration of charge delocalization phenomena, and both metal-based as well as purely organic redox-active units have been used for this purpose.6 A considerable number of thiophene-bridged mixed valence compounds have been studied,7 but selenophene, furan, and tellurophene bridges have received little to no attention.7k,8 A very recent study reported that oligofurans can mediate stronger electronic coupling between ferrocenyl redox units than oligothiophenes.8a We are unaware of prior comparative studies of charge delocalization encompassing the entire chalcogenophene series (Scheme 1).
 | ||
| Scheme 1 Molecular structure of the systems investigated in this study. | ||
Our study is based on four isostructural compounds with phenothiazine (PTZ) groups attached at the 2- and 5-positions of furan (1), thiophene (2), selenophene (3), and tellurophene (4). An X-ray crystal structure of the selenophene compound is shown in Fig. 1. The structure of the thiophene analogue (2) had been previously reported.7m Synthetic procedures and product characterization data are given in the ESI.†
 | ||
| Fig. 1 X-ray crystal structure of compound 3. | ||
Cyclic voltammetry reveals two essentially reversible oxidation waves in the potential range between 0 and 0.6 V vs. Fc+/Fc for all four compounds (Fig. 2), corresponding to consecutive one-electron oxidation of both PTZ moieties. Chalcogenophene-based oxidations are expected at much higher potentials.9 In CH3CN with 0.1 M TBAPF6 the splitting between peak potentials (ΔE) increases along the chalcogenophene series, ranging from 216 mV for furan to 291 mV for tellurophene (Table 1). Using these ΔE values one calculates comproportionation constants (Kc = 10ΔE/59mV) between 4.5 × 103 and 8.6 × 104 in CH3CN (Table 1). In general, ΔE and Kc are governed by several factors including electrostatic and ion pairing effects, as well as the strength of electronic interaction (HAB) between the two redox centers.10 Electrostatic and ion pairing effects often dominate, and therefore conclusions about HAB based on electrochemical data should be made with caution.8b In the present case the observed increase of ΔE and Kc along the series of chalcogenophenes 1–4 is difficult to reconcile with electrostatics because the distance between redox centers is not decreasing along this series.
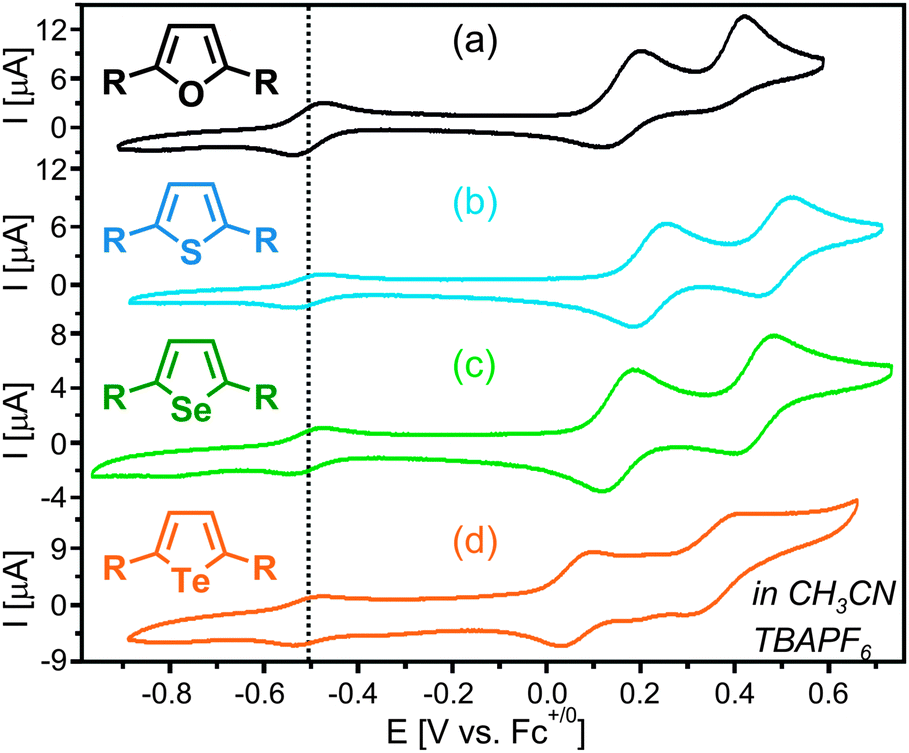 | ||
| Fig. 2 Cyclic voltammograms recorded in CH3CN with 0.1 M TBAPF6. The scan rate was 100 mV s−1. | ||
| Compd | ΔE [mV] | K c |
|---|---|---|
| 1 | 216 | 4.5× 103 |
| 2 | 267 | 3.4× 104 |
| 3 | 283 | 6.3× 104 |
| 4 | 291 | 8.6× 104 |
While 4 × 10−5 M solutions of the charge-neutral forms of 1–4 in CH3CN are essentially colorless (dotted black traces in Fig. 3), oxidation with Cu(ClO4)2 produces new absorptions in the near-infrared and visible spectral ranges. In all four cases the lowest-energy absorption band is centered around 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–15000 cm−1, reaching its maximum intensity after addition of 1 equivalent of Cu(II) oxidant (red traces, bands marked with asterisks) from which we conclude that these bands are due to the mixed-valent 1+–4+ species. As frequently observed, addition of a second equivalent of Cu(ClO4)2 oxidant is unable to quantitatively convert the mono- to the dications.6f However, the abovementioned low-energy bands clearly lose intensity when more than 1 equivalent of oxidant is added, and for 22+–42+ a new absorption band around 19500 cm−1 becomes detectable (dash-dotted black traces). In the case of molecule 1 addition of a second equivalent of oxidant rapidly leads to sample decomposition.
500–15000 cm−1, reaching its maximum intensity after addition of 1 equivalent of Cu(II) oxidant (red traces, bands marked with asterisks) from which we conclude that these bands are due to the mixed-valent 1+–4+ species. As frequently observed, addition of a second equivalent of Cu(ClO4)2 oxidant is unable to quantitatively convert the mono- to the dications.6f However, the abovementioned low-energy bands clearly lose intensity when more than 1 equivalent of oxidant is added, and for 22+–42+ a new absorption band around 19500 cm−1 becomes detectable (dash-dotted black traces). In the case of molecule 1 addition of a second equivalent of oxidant rapidly leads to sample decomposition.
 | ||
| Fig. 3 Absorption spectra recorded in absence and in presence of various amounts of Cu(ClO4)2 oxidant in CH3CN. The asterisks mark the IVCT band. | ||
By analogy to many previously investigated bis(triarylamines) and related organic mixed valence compounds we associate the low-energy absorptions of 1+–4+ (marked by the red asterisks) with intervalence charge transfer (IVCT) bands.6b,f,h,7g,j,k,p,s,11 The optical absorption spectra with properly determined extinction coefficients were fitted to a sum of Gaussian functions in order to estimate the dipole moment (μge) associated with the IVCT transition (Fig. S1 in the ESI†). Eqn (1) requires νmax (the energetic position of the absorption band maximum) and ε(ν) input values in units of cm−1 and M−1 cm−1, respectively, and yields the transition dipole moment in units of Debyes (D).12
 | (1) |
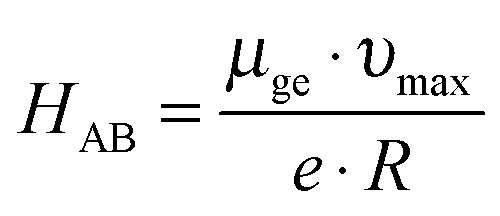 | (2) |
In eqn (2), e is the elemental charge and R is the effective charge transfer distance. In compounds 1–4 the N–N distance is expected to vary between 4.58 and 5.32 Å based on molecular modeling and X-ray crystal structure data (Table 1). Prior studies of mixed-valent bis(triarylamine) cations and dinitroaromatic anions have reached the conclusion that R is equal to roughly 2/3 of the geometrical N–N distance (dNN) in these systems.14 Therefore it seems plausible to assume that R ≈ 2/3·dNN for 1+–4+, and this leads to estimates for the electronic coupling matrix elements in the range from ∼3300 to ∼4100 cm−1 (Table 2). For comparison, the structurally related N,N,N′,N′-tetraanisyl-p-phenylenediamine cation has HAB = 3240 cm−1.11a The solvent dependence of the IVCT band is relatively weak, νmax red-shifts between ∼500 and ∼1500 cm−1 when changing from CH3CN to CH2Cl2 (Fig. S2, ESI†).
The EPR spectra of 1+–4+ in CH3CN indicate that the unpaired electron spin interacts with the nuclear spins of 14N and 1H (solid traces in Fig. 4). Simulation of the experimental spectra (dotted traces in Fig. 4) yields the gyromagnetic factors g, and the hyperfine coupling constants reported in Table 3. All EPR spectra are centered at values of the gyromagnetic factors g ranging from 2.0024 to 2.0032, typical for organic radicals. The simulations indicate an interaction of the unpaired electron with two equivalent nitrogen nuclei (aN ranging from 4.2 to 5.2 G), and two equivalent hydrogen nuclei (aH varying from 3.6 to 4.8 G) for the two chalcogenophene H-atoms. Nitrogen hyperfine coupling constants (aN) between 4.2 and 5.2 G for 1+–4+ are compatible with complete delocalization of the unpaired electron over two N nuclei on the EPR timescale. In the case of 4+ the presence of an impurity somewhat complicates analysis of the EPR data (see ESI† for details) but reliable parameters can nevertheless be determined. For 1+–3+ there is no indication for hyperfine interaction with the chalcogenophene heteroatom. For 4+ interaction of the unpaired electron with the tellurium atom cannot be rigorously excluded (see ESI†).
 | ||
| Fig. 4 Solid lines: experimental X-band EPR spectra recorded for the monocationic forms in CH3CN. Dotted lines: simulated spectra. See ESI.† | ||
| Compd | g | a N [G] | a H [G] |
|---|---|---|---|
| 1+ | 2.0032 | 4.2 | 4.8 |
| 2+ | 2.0032 | 4.8 | 3.9 |
| 3+ | 2.0024 | 4.9 | 3.6 |
| 4+ | 2.0030 | 5.2 | 4.0 |
The combined data sets from cyclic voltammetry, optical absorption, and EPR spectroscopy are in line with strong charge delocalization in 1+–4+. ΔE and Kc increase systematically along the chalcogenophene series (Table 1), but for μge and HAB this trend is not followed (Table 2). The optical absorption data rather suggests that furan and tellurophene mediate electronic coupling somewhat less well than thiophene and selenophene, but the differences are relatively small when considering the uncertainty associated with the procedure used for determination of μge and HAB. We are therefore lead to the conclusion that single units of furan, thiophene, selenophene, and tellurophene mediate electronic coupling between two amine redox centers similarly well. This is consistent with a picture in which the electronic communication between the 2- and 5-positions of a chalcogenophene is mostly mediated by the butadiene backbone of the heterocycle, and the EPR data is compatible with this view. A recent study of ferrocene-based mixed valence compounds has reached the same conclusion for phosphole bridges.15
We have conducted the first comparative study of charge delocalization across single units of the entire chalcogenophene series, and our findings are relevant in the greater context of molecular electronics,16 for instance for the design of new chalcogenophene-based charge-conducting oligomers and polymers.
Funding from the Swiss NSF through grant number 200021_146231/1 is gratefully acknowledged. CGP acknowledges the Swiss NSF for a R'Equip grant for the EPR spectrometer.
Notes and references
- (a) X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed; (b) J. L. Brédas, J. P. Calbert, D. A. da Silva and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- (a) J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS; (b) T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
- (a) E. Poverenov, Y. Sheynin, N. Zamoshchik, A. Patra, G. Leitus, I. F. Perepichka and M. Bendikov, J. Mater. Chem., 2012, 22, 14645–14655 RSC; (b) A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422–433 RSC; (c) Y. H. Wijsboom, A. Patra, S. S. Zade, Y. Sheynin, M. Li, L. L. W. Shimon and M. Bendikov, Angew. Chem., Int. Ed., 2009, 48, 5443–5447 CrossRef CAS PubMed; (d) M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC.
- (a) O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC; (b) O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed; (c) A. A. Jahnke and D. S. Seferos, Macromol. Rapid Commun., 2011, 32, 943–951 CrossRef CAS PubMed; (d) A. A. Jahnke, G. W. Howe and D. S. Seferos, Angew. Chem., Int. Ed., 2010, 49, 10140–10144 CrossRef CAS PubMed.
- (a) O. Gidron, Y. Diskin-Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148–2149 CrossRef CAS PubMed; (b) A. A. Jahnke, B. Djukic, T. M. McCormick, E. B. Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed; (c) C. R. B. Rhoden and G. Zeni, Org. Biomol. Chem., 2011, 9, 1301–1313 RSC.
- (a) W. Kaim, A. Klein and M. Glöckle, Acc. Chem. Res., 2000, 33, 755–763 CrossRef CAS PubMed; (b) S. F. Nelsen, Chem. – Eur. J., 2000, 6, 581–588 CrossRef CAS; (c) K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed; (d) D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed; (e) F. Pevny, E. Di Piazza, L. Norel, M. Drescher, R. F. Winter and S. Rigaut, Organometallics, 2010, 29, 5912–5918 CrossRef CAS; (f) J. Hankache and O. S. Wenger, Chem. Rev., 2011, 111, 5138–5178 CrossRef CAS PubMed; (g) D. Siebler, M. Linseis, T. Gasi, L. M. Carrella, R. F. Winter, C. Forster and K. Heinze, Chem. – Eur. J., 2011, 17, 4540–4551 CrossRef CAS PubMed; (h) A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed; (i) P. J. Low, Coord. Chem. Rev., 2013, 257, 1507–1532 CrossRef CAS PubMed.
- (a) J. Guay, P. Kasai, A. Diaz, R. L. Wu, J. M. Tour and L. H. Dao, Chem. Mater., 1992, 4, 1097–1105 CrossRef CAS; (b) P. Bäuerle, U. Segelbacher, A. Maier and M. Mehring, J. Am. Chem. Soc., 1993, 115, 10217–10223 CrossRef; (c) P. Bäuerle, U. Segelbacher, K. U. Gaudl, D. Huttenlocher and M. Mehring, Angew. Chem., Int. Ed., 1993, 32, 76–78 CrossRef; (d) Y. B. Zhu and M. O. Wolf, J. Am. Chem. Soc., 2000, 122, 10121–10125 CrossRef CAS; (e) S. Le Stang, F. Paul and C. Lapinte, Organometallics, 2000, 19, 1035–1043 CrossRef CAS; (f) S. Fraysse, C. Coudret and J.-P. Launay, J. Am. Chem. Soc., 2003, 125, 5880–5888 CrossRef CAS PubMed; (g) D. Rohde, L. Dunsch, A. Tabet, H. Hartmann and J. Fabian, J. Phys. Chem. B, 2006, 110, 8223–8231 CrossRef CAS PubMed; (h) J. C. Lacroix, K. I. Chane-Ching, F. Maquère and F. Maurel, J. Am. Chem. Soc., 2006, 128, 7264–7276 CrossRef CAS PubMed; (i) P. Rapta, O. Zeika, D. Rohde, H. Hartmann and L. Dunsch, ChemPhysChem, 2006, 7, 863–870 CrossRef CAS PubMed; (j) S. A. Odom, K. Lancaster, L. Beverina, K. M. Lefler, N. J. Thompson, V. Coropceanu, J. L. Brédas, S. R. Marder and S. Barlow, Chem. – Eur. J., 2007, 13, 9637–9646 CrossRef CAS PubMed; (k) G. Nöll, M. Avola, M. Lynch and J. Daub, J. Phys. Chem. C, 2007, 111, 3197–3204 CrossRef; (l) L. B. Gao, J. Kan, Y. Fan, L. Y. Zhang, S. H. Liu and Z. N. Chen, Inorg. Chem., 2007, 46, 5651–5664 CrossRef CAS PubMed; (m) A. W. Franz, L. N. Popa, F. Rominger and T. J. J. Müller, Org. Biomol. Chem., 2009, 7, 469–475 RSC; (n) J. Casado, S. R. Gonzalez, M. C. R. Delgado, M. M. Oliva, J. T. L. Navarrete, R. Caballero, P. de la Cruz and F. Langa, Chem. – Eur. J., 2009, 15, 2548–2559 CrossRef CAS PubMed; (o) F. Zhang, G. Gotz, E. Mena-Osteritz, M. Weil, B. Sarkar, W. Kaim and P. Bäuerle, Chem. Sci., 2011, 2, 781–784 RSC; (p) S. Barlow, C. Risko, S. A. Odom, S. J. Zheng, V. Coropceanu, L. Beverina, J. L. Brédas and S. R. Marder, J. Am. Chem. Soc., 2012, 134, 10146–10155 CrossRef CAS PubMed; (q) J. M. Speck, R. Claus, A. Hildebrandt, T. Rüffer, E. Erasmus, L. van As, J. C. Swarts and H. Lang, Organometallics, 2012, 31, 6373–6380 CrossRef CAS; (r) Y. P. Ou, J. L. Xia, J. Zhang, M. Xu, J. Yin, G. A. Yu and S. H. Liu, Chem. – Asian J., 2013, 8, 2023–2032 CrossRef CAS PubMed; (s) L. G. Reuter, A. G. Bonn, A. C. Stückl, B. C. He, P. B. Pati, S. S. Zade and O. S. Wenger, J. Phys. Chem. A, 2012, 116, 7345–7352 CrossRef CAS PubMed; (t) M. Jenart, C. Niebel, J. Y. Balandier, J. Leroy, A. Mignolet, S. Stas, A. Van Vooren, J. Cornil and Y. H. Geerts, Tetrahedron, 2012, 68, 349–355 CrossRef CAS PubMed.
- (a) O. Gidron, Y. Diskin-Posner and M. Bendikov, Chem. – Eur. J., 2013, 19, 13140–13150 CrossRef CAS PubMed; (b) D. M. D'Alessandro and F. R. Keene, Dalton Trans., 2004, 3950–3954 RSC.
- (a) H. Nakanishi, S. Inoue and T. Otsubo, Mol. Cryst. Liq. Cryst., 1997, 296, 335–348 CrossRef CAS; (b) S. Inoue, H. Nakanishi, K. Takimiya, Y. Aso and T. Otsubo, Synth. Met., 1997, 84, 341–342 CrossRef CAS.
- A. Hildebrandt and H. Lang, Organometallics, 2013, 32, 5640–5653 CrossRef CAS.
- (a) C. Lambert and G. Nöll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS; (b) C. Lambert, G. Nöll and J. Schelter, Nat. Mater., 2002, 1, 69–73 CrossRef CAS PubMed; (c) S. Barlow, C. Risko, S. J. Chung, N. M. Tucker, V. Coropceanu, S. C. Jones, Z. Levi, J. L. Brédas and S. R. Marder, J. Am. Chem. Soc., 2005, 127, 16900–16911 CrossRef CAS PubMed; (d) J. Bonvoisin, J. P. Launay, M. Van der Auweraer and F. C. De Schryver, J. Phys. Chem., 1994, 98, 5052–5057 CrossRef CAS; (e) D. Sun, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2005, 44, 5133–5136 CrossRef CAS PubMed.
- (a) C. Creutz, M. D. Newton and N. Sutin, J. Photochem. Photobiol., A, 1994, 82, 47–59 CrossRef CAS; (b) R. J. Cave and M. D. Newton, Chem. Phys. Lett., 1996, 249, 15–19 CrossRef CAS.
- N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391 CrossRef CAS.
- (a) S. F. Nelsen, A. E. Konradsson, M. N. Weaver and J. P. Telo, J. Am. Chem. Soc., 2003, 125, 12493–12501 CrossRef CAS PubMed; (b) S. F. Nelsen, M. N. Weaver, J. I. Zink and J. P. Telo, J. Am. Chem. Soc., 2005, 127, 10611–10622 CrossRef CAS PubMed; (c) K. Lancaster, S. A. Odom, S. C. Jones, S. Thayumanavan, S. R. Marder, J. L. Brédas, V. Coropceanu and S. Barlow, J. Am. Chem. Soc., 2009, 131, 1717–1723 CrossRef CAS PubMed.
- D. Miesel, A. Hildebrandt, M. Korb, P. J. Low and H. Lang, Organometallics, 2013, 32, 2993–3002 CrossRef CAS.
- C. Herrmann and J. Elmisz, Chem. Commun., 2013, 49, 10456–10458 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis protocols and product characterization data, additional optical spectroscopic and EPR data. CCDC 1002373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03806a |
| This journal is © The Royal Society of Chemistry 2014 |