 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly regioselective lithiation of pyridines bearing an oxetane unit by n-butyllithium†
Guy
Rouquet
*a,
David C.
Blakemore
b and
Steven V.
Ley
a
aDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: gr354@cam.ac.uk
bNeusentis Chemistry, Pfizer Worldwide Research and Development, The Portway Building, Granta Park, Cambridge, CB21 6GS, UK
First published on 20th June 2014
Abstract
The first regioselective ortho-lithiation at the 4-position of simple pyridine derivatives containing a 3-oxetane unit has been achieved using n-butyllithium as base. Electrophilic quenching of the resulting lithio species provides a rapid access to a broad range of new functionalized pyridine oxetane building blocks.
The oxetane ring is an intriguing and key structural motif present in a number of biologically active natural products1e such as taxol,2 antibiotic oxetin3 or antiviral agent oxetanocin.4 Neglected for a long time, primarily due to concerns about chemical and metabolic instability, oxetanes are currently subject to an increasing interest in medicinal chemistry as they have been found to be a useful unit for improving the physicochemical and biochemical properties of drug candidates.1 In a wider context, the use of oxetanes can also be extended to agrochemical,1e,5 polymer6a–c and material sciences.6d,e As immediate higher analogues of epoxides, oxetanes are also powerful synthetic intermediates, offering interesting new synthetic disconnections via ring opening and rearrangement reactions.1e,7 Traditional approaches to the synthesis of oxetanes include the Williamson ether synthesis,8 the Paternò–Büchi [2+2] cycloaddition9 and the one-pot addition of sulfoxonium ylides to aldehydes or ketones.10 Although these methods allow access to a large range of oxetanes, the preparation of elaborated oxetanes bearing a higher degree of functionality is often laborious and can represent a significant synthetic challenge. Therefore, despite their promising potential, the study and exploitation of oxetane containing molecules remains underdeveloped.
Recently, Capriati has shown that 2-aryloxetanes could be functionalized regioselectivity via an ortho-lithiation reaction using s-BuLi as a base, opening a new dimension for the preparation of oxetanes (eqn (1), Scheme 1).11 This new directed ortho-metallation reaction (DoM)12 was rationalized through the ability of the oxetane ring to act as a directed metallation group (DMG).12
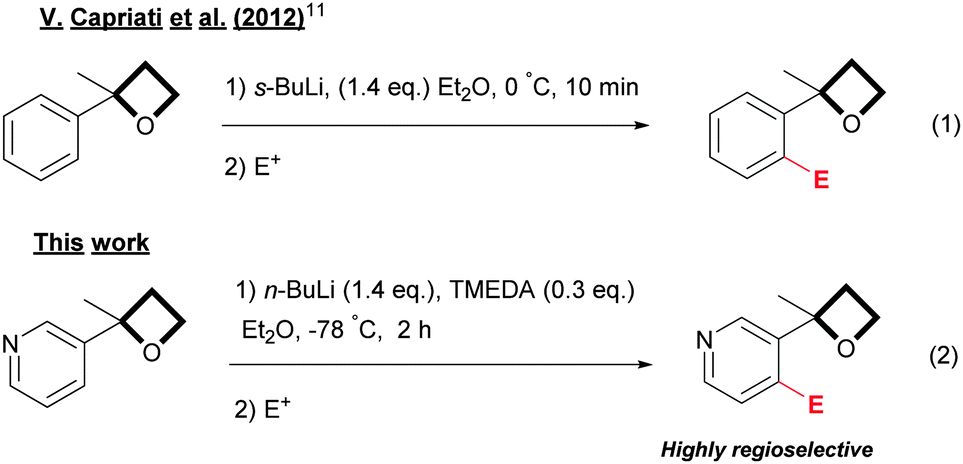 | ||
| Scheme 1 ortho-Lithiation directed by an oxetane ring. | ||
Heterocyclic derivatives are of considerable importance as scaffolds in drug discovery. The pyridine ring is particularly attractive13 as it is much less lipophilic than a phenyl ring and relatively electron-poor (electron-rich heterocycles are prone to oxidation). Accordingly, extending this reaction to pyridine oxetanes is appealing. Nevertheless, the ortho-lithiation of pyridines14 is known to be quite difficult and complex in comparison with its benzene counterpart, due to the presence of the nitrogen atom which can dramatically affect the reactivity toward organolithium reagents.
Herein, we report that 3-oxetane pyridines can be efficiently ortho-functionalized in a regioselective manner at the 4-position under straightforward conditions by using n-BuLi as a base in combination with a sub-stoichiometric amount of TMEDA (eqn (2), Scheme 1).
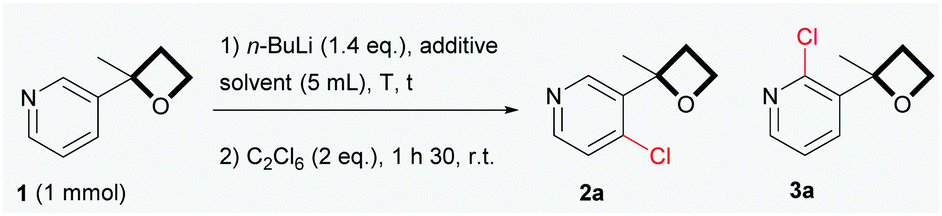
In our preliminary investigations (Table 1), the ortho-chlorination of 3-(2-methyloxetan-2-yl)pyridine 1, prepared in large scale through the one-pot addition of sulfoxonium ylides to 3-acetylpyridine,15 was chosen as a test reaction. Not surprisingly, by using n-BuLi under reaction conditions similar to those reported by Capriati for the lithiation of aryl derivatives (eqn (1), Scheme 1), a complex mixture, containing mainly various alkylation and ring opening products, was obtained (entry 1).16 However, an in-depth analysis of the NMR 1H revealed the presence of a small amount of chlorinated products 2a and 3a, which encouraged us to persevere in developing the reaction. Gratifyingly, we found that the simple addition of 1.4 equivalent of TMEDA (entry 2) had a profound impact on the progress of the reaction,17 providing a cleaner and readily characterizable crude reaction mixture. Regioisomers 2a and 3a were isolated in respective yields of 46% and 15% after flash chromatography over silica gel. Reaction temperatures were also a key issue in the control of the reactivity. Lowering the reaction temperatures considerably diminished the formation of unwanted side products, which were totally suppressed when the reaction was carried out at −78 °C (entries 3 and 4). However, at −78 °C, the reaction proved to be relatively slow (entries 4 and 5) and driving the conversion of 1, by increasing the reaction times or the number of equivalents of base used, led to increased side-products being formed. Based on our preliminary observations concerning the positive effect of TMEDA (entry 2), we found that the addition of 1.4 equivalent of TMEDA at −78 °C (entry 7) dramatically increased reaction-efficiency to finally afford a very clean reaction giving 2a in a yield of 88%, accompanied by the regioisomer 3a as a minor product (4%). To our delight, when a sub-stoichiometric amount of TMEDA was used (entry 8), almost no product 3a was formed (trace amounts were detected by GC/MS),18 while maintaining a satisfactory yield of 2a. To ensure a full and reproducible conversion of 1, a reaction time of 2 h was advisable (based on NMR 1H, less than 5% of the starting material remains after 1 h 30). As anticipated, lower yields were obtained when THF was used as solvent (entry 9).19 Our attempts to deprotonate 1 with LDA were unprofitable.20 Poor yields were obtained and we faced a high propensity for the competitive ring opening of 116 at temperature below −78 °C. The non-nucleophilic TMSCH2Li base has proven to be relatively efficient21 for this transformation and generated 2a as an exclusive product in a yield of 75%. Although TMSCH2Li can be considered as a viable alternative, n-BuLi was the base of choice (particularly for scale-up applications) considering the high cost and lower availability of TMSCH2Li.
|
|
|||||
|---|---|---|---|---|---|
| Entry | t (h) | T (°C) | Solvent | Additive | 2a/3a/1a,b (%) |
| a Recovered 1. b Yields isolated by column chromatography on silica gel. c 1.4 equivalent were used. d With prolonged reaction times, decomposition products appeared. e 0.3 equivalent were used. f Trace amount of 3a was detected by GC/MS. | |||||
| 1 | 1.5 | 0 | Et2O | None | Complex mixture |
| 2 | 1.5 | 0 | Et2O | TMEDAc | 46/15/0d |
| 3 | 1.5 | −30 | Et2O | None | 34/4/0 |
| 4 | 1.5 | −78 | Et2O | None | 23/0/64 |
| 5d | 6 | −78 | Et2O | None | 55/0/32 |
| 6 | 1.5 | −30 | Et2O | TMEDAc | 63/6/0 |
| 7 | 1.5 | −78 | Et2O | TMEDAc | 88/4/0 |
| 8 | 2 | −78 | Et2O | TMEDAe | 85/0f/0 |
| 9 | 2 | −78 | THF | TMEDAe | 64/0f/0 |
With these satisfactory reaction conditions in hand, we then examined the flexibility of the reaction with various representative electrophiles (Scheme 2). The resulting anion was quenched with trimethylsilyl chloride and phenyl disulfide, providing 2b and 2c respectively in yields of 78 and 91%. In addition to the chlorinated 2a, brominated and iodinated compounds 2d and 2e were smoothly obtained in yields of 82 and 79%. By quenching with DMF, phenyl isocyanate and ethyl cyano formate, various C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups were also incorporated with success and aldehydes 2f, amide 2g and ester 2h were isolated in a good yield. Addition to aldehydes has also proven to be effective, offering aliphatic and aromatic alcohols 2i and 2j as diastereomers. The reaction was then extended to imino acceptors with a N-benzylsulfonyl imine and a diphenylimine, affording the corresponding amines 2k and 2l in a moderate yield. Interestingly, a relatively selective diastereomeric ratio was observed, suggesting potential perspectives for further applications in diastereo controlled synthesis.
O functional groups were also incorporated with success and aldehydes 2f, amide 2g and ester 2h were isolated in a good yield. Addition to aldehydes has also proven to be effective, offering aliphatic and aromatic alcohols 2i and 2j as diastereomers. The reaction was then extended to imino acceptors with a N-benzylsulfonyl imine and a diphenylimine, affording the corresponding amines 2k and 2l in a moderate yield. Interestingly, a relatively selective diastereomeric ratio was observed, suggesting potential perspectives for further applications in diastereo controlled synthesis.
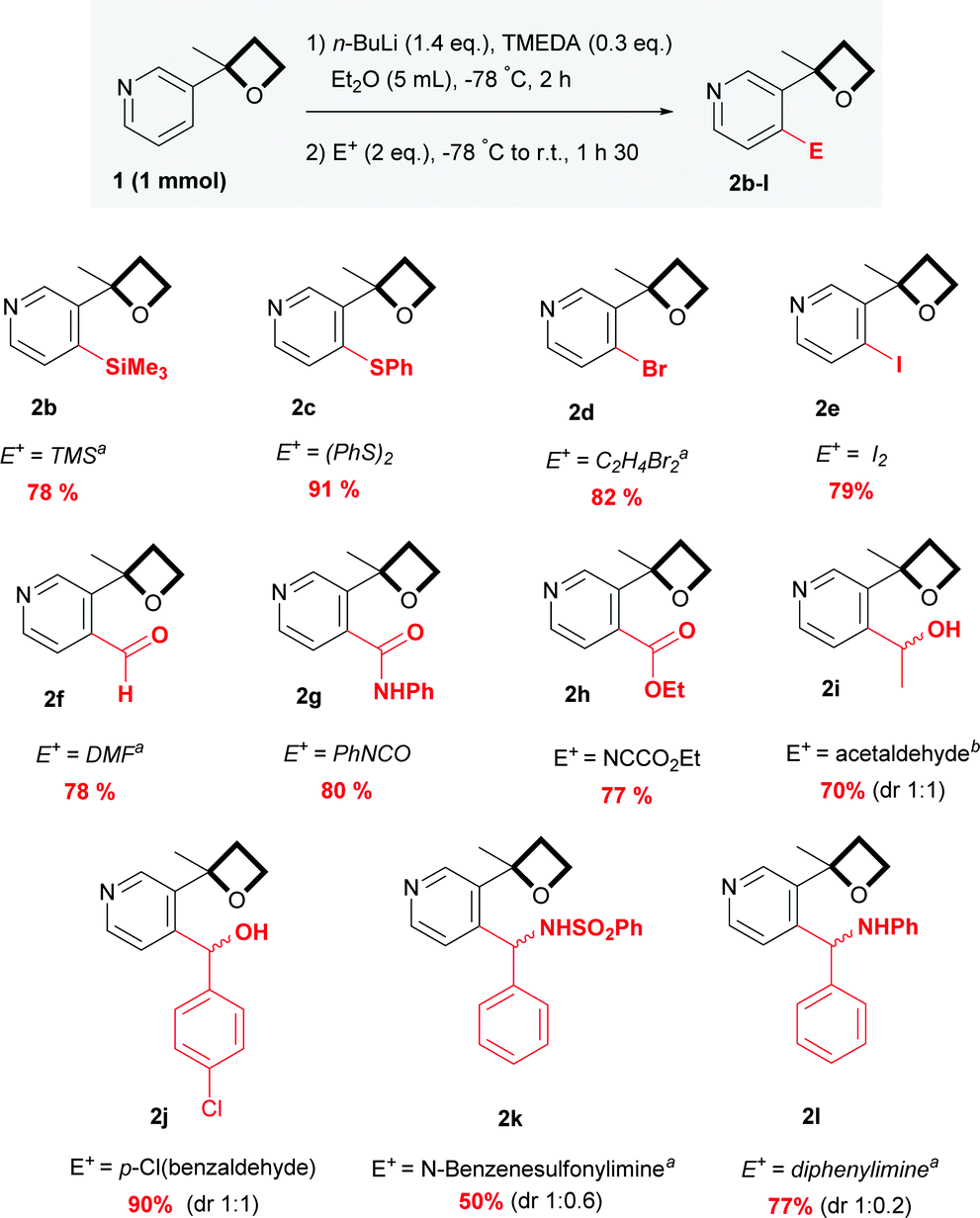 | ||
| Scheme 2 ortho-Functionalization of 1. a 3 eq. were used. b 5 eq. were used. | ||
According to the conclusions of Capriati, the oxetane should be ranked as a moderately strong directing group,22 which allows the presence on the aromatic ring of functional groups known to be weak directing groups without altering the regioselectivity of the reaction.11 In line with these observations, oxetane pyridines 5 and 6, bearing respectively a chloro and a methoxy group, reacted smoothly with a complete regioselectivity at the site adjacent to the oxetane unit (Scheme 3, eqn (3)). As reported for the functionalization of 2-aryl oxetane,11 variation on the α-position of the 3-pyridine oxetane is also tolerated, indeed oxetane 7 where the methyl group was substituted by an ethyl was successfully functionalized by quenching with DMF, providing aldehyde 10 in a good yield (Scheme 3, eqn (4)).
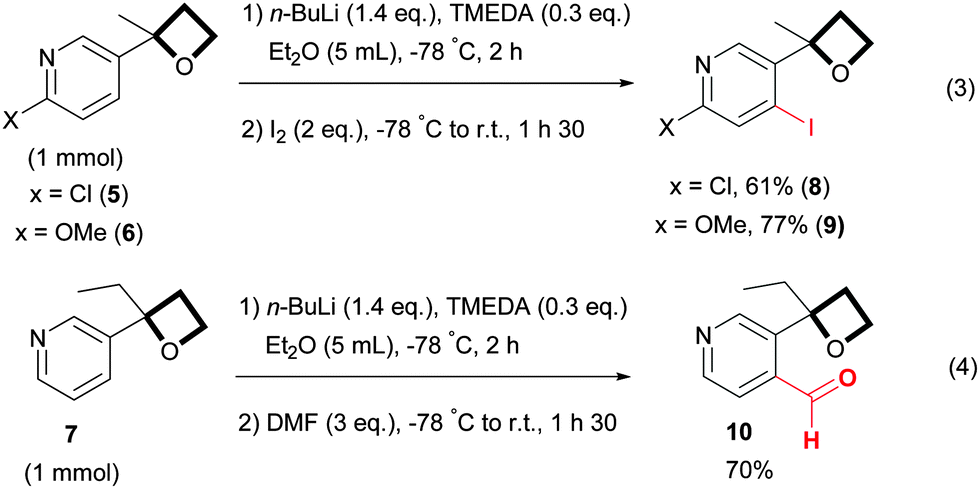 | ||
| Scheme 3 ortho-Lithiation of various 3-oxetane pyridines. | ||
Deuterium labelling experiments with methanol-D4 as a deuterium quenching source (Scheme 4, eqn (5)) confirmed the high regioselectivity and excellent conversion rates of the reaction. Significant amounts of the D-labelled C-2 and C-3 regioisomers were not detectable by NMR 1H under those reaction conditions. Quéguiner,23 throughout his pioneering studies, assumed that in some cases, when alkyl lithium bases are used, chelation between the pyridine nitrogen and the lithium may take place, enhancing the acidity of the 2-proton of the pyridine and thus facilitating its direct abstraction via an inductive effect.24 It has been observed that slow equilibration can then occur which results in a translocation of the C-2 anion to the position C-4. At first sight it seems unlikely that such a process occurred in the present reaction. Indeed, quenching of the reaction after 3 min reaction time with methanol-D4 (Scheme 4, eqn (6)) showed an exclusive incorporation of deuterium at the 4-position. The formation of C-4 regioisomers, thus, likely results from an initial abstraction of the C-4 proton via a DoM reaction and this is in accord with Capriati's finding regarding the DMG ability of the oxetane motif for the ortho lithiation of 2-aryl oxetanes.11 In addition, in pyridine itself, the relative acidities of the proton at the 4-, 3-, and 2-positions are 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :72:1.25 The position next to the nitrogen is commonly considered least acidic because the sp2 lone pair on nitrogen has an anti-bonding interaction with the C–Li bond.23b,26 The regioselectivity observed in the present reaction is consistent with this.
:72:1.25 The position next to the nitrogen is commonly considered least acidic because the sp2 lone pair on nitrogen has an anti-bonding interaction with the C–Li bond.23b,26 The regioselectivity observed in the present reaction is consistent with this.
 | ||
| Scheme 4 Deuterium labelling experiments. | ||
The scalability of the reaction was then examined. The ortho-functionalization of 1 was conducted safely on a gram-scale (Scheme 5, eqn (7)) in methyl tert-butyl ether (MTBE), which has been found to be an equally suitable solvent for this reaction. We finally considered the possibility of achieving further elaborations through metal-catalyzed couplings. Under typical Suzuki cross-coupling conditions, iodide 2e reacted smoothly with various arylboronic acids (Scheme 5, eqn (8)), affording expected arylation products 11, 12 and 13 in good yields.
 | ||
| Scheme 5 Gram-scale ortho-lithiation of 1 and Suzuki cross-coupling of 2e. | ||
In conclusion, the ability of the oxetane ring to promote the ortho-lithiation of 3-oxetane pyridine derivatives has been described for the first time and a variety of novel C-4 functionalized pyridine oxetanes have been synthesized through this strategy.27 The present method offers good yields, a remarkably high regioselectivity and provides a rapid and cheap access to new valuable functionalized building blocks from readily available oxetane precursors.28 Ongoing research is underway in our laboratory to extend the methodology to other N-heterocycles containing an oxetane motif and to develop their exploitation in the synthesis of compounds of pharmaceutical interest.
We are grateful to Pfizer Worldwide Research and Development (G.R.) and the BP endowment (S.V.L.) for financial support.
Notes and references
- Selected publications: (a) G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736 CrossRef CAS PubMed; (b) G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512 CrossRef CAS PubMed; (c) A. F. Stepan, K. Karki, W. S. McDonald, P. H. Dorff, J. K. Dutra, K. J. DiRico, A. Won, C. Subramanyam, I. V. Efremov, C. J. O'Donnell, C. E. Nolan, S. L. Becker, L. R. Pustilnik, B. Sneed, H. Sun, Y. Lu, A. E. Robshaw, D. Riddell, T. J. O'Sullivan, E. Sibley, S. Capetta, K. Atchison, A. J. Hallgren, E. Miller, A. Wood and R. S. Obach, J. Med. Chem., 2011, 54, 7772 CrossRef CAS PubMed; (d) J. Du, B.-K. Chun, R. T. Mosley, S. Bansal, H. Bao, C. Espiritu, A. M. Lam, E. Murakami, C. Niu, H. M. M. Steuer, P. A. Furman and M. J. Sofia, J. Med. Chem., 2014, 57, 1826 CrossRef CAS PubMed ; for key reviews, see: ; (e) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed; (f) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227 CrossRef CAS PubMed.
- K. C. Nicolaou and R. K. Guy, Angew. Chem., Int. Ed., 1995, 34, 2079 CrossRef CAS.
- Y. Kawahata, S. Takatsuto, N. Ikekawa, M. Murata and S. Ōmura, Chem. Pharm. Bull., 1986, 34, 3102 CrossRef CAS.
- D. W. Norbeck and J. B. Kramer, J. Am. Chem. Soc., 1988, 110, 7217 CrossRef CAS.
- Selected publications: (a) G. Holan, Nature, 1971, 232, 644 CrossRef CAS; (b) W. Meyer, Ciba-Geigy AG, EP 92–810027, 1992 Search PubMed.
- Selected publications: (a) J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4331 CrossRef CAS; (b) Y. Permana, K. Nakano, M. Yamashita, D. Watanabe and K. Nozaki, Chem. – Asian J., 2008, 3, 710 CrossRef CAS PubMed; (c) H. Bouchékif, M. I. Philbin, E. Colclough and A. J. Amass, Macromolecules, 2008, 41, 1989 CrossRef; (d) C. Ulbricht, N. Rehmann, E. Holder, D. Hertel, K. Meerholz and U. S. Schubert, Macromol. Chem. Phys., 2009, 210, 531 CrossRef CAS; (e) A. Charas and J. Morgado, CurrPhysChem, 2012, 2, 241 CAS.
- Recent representative examples: (a) B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675 CrossRef CAS PubMed; (b) W. Zhao, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 6209 CrossRef CAS PubMed; (c) S. A. Ruider, S. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11908 CrossRef CAS PubMed.
- T. Aftab, C. Carter, M. Christlieb, J. Hart and A. Nelson, J. Chem. Soc., Perkin Trans. 1, 2000, 711 RSC.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed.
- K. Okuma, Y. Tanaka, S. Kaji and H. Ohta, J. Org. Chem., 1983, 48, 5133 CrossRef CAS.
- D. I. Coppi, A. Salomone, F. M. Perna and V. Capriati, Angew. Chem., Int. Ed., 2012, 51, 7532 CrossRef CAS PubMed.
- V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS.
- (a) M. D. Hill, Chem. – Eur. J., 2010, 16, 12052 CrossRef CAS PubMed; (b) A. Chaubey and S. N. Pandeya, Asian J. Pharm. Clin. Res., 2011, 4, 5 CAS; (c) J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, 5th edn, 2010 Search PubMed; (d) A. E. Goetz and N. K. Garg, Nat. Chem., 2012, 5, 54 CrossRef PubMed; (e) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014 DOI:10.1021/jm4017625.
- For an overview: (a) F. Mongin and G. Quéguiner, Tetrahedron, 2001, 57, 4059 CrossRef CAS; (b) G. W. Gribble, Science of Synthesis, Georg thieme Verlag KG, 2006, Section 8.1.14, p. 395 Search PubMed.
- See ESI† for the experimental procedure.
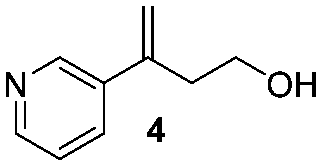
- The alcohol 4 was isolated. We assumed that 4 was the product of an opening of the oxetane ring induced by the base (see Introduction of ref. 11)
. - For the role of TMEDA in lithiation reactions, see for example: D. B. Collum, Acc. Chem. Res., 1992, 25, 448 CrossRef CAS.
- According to GC/MS less than 0.5% of 3a was formed.
- It has been supposed that THF may partially replace the oxetane ring in coordinating to lithium, which could have a negative impact on the reaction (see ref. 11). Formation of an undefined mixture of background products was also observed.
- LDA is likely not basic enough to efficiently deprotonate 1, see for example: D. L. Comins and D. H. LaMunyon, Tetrahedron Lett., 1988, 29, 773 CrossRef CAS.
- Reaction conditions: 1 mmol of 1 with 1.5 eq. of TMSCH2Li, 1.5 eq. of TMEDA, at −78 °C for 2 h in Et2O (5 mL). For recent reports of the use of TMSCH2Li in ortho-lithiation reactions: (a) C. J. Woltermann and D. E. Sutton, WO2004/092125A2, 2004; (b) P. C. Gros, A. Doudouh and C. Woltermann, Org. Biomol. Chem., 2006, 4, 4331 RSC.
- The oxetane is almost as strong as a dimethylaminomethyl substituent to direct ortho-lithiation (see ref. 11).
- (a) F. Marsais, G. Le Nard and G. Quéguiner, Synthesis, 1982, 235 CrossRef CAS; (b) F. Marsais and G. Quéguiner, Tetrahedron, 1983, 39, 2009 CrossRef CAS.
- That the process occurred to such a small extent may explain why a very small quantity of the C-2 regioisomer was formed (see Table 1). A direct abstraction assisted by the oxetane (DoM) may also be considered.
- J. Clayden, Organolithiums: Selectivity for synthesis, Elsevier Science Ltd, Oxford, UK, 2002 Search PubMed.
- V. L. Blair, D. C. Blakemore, D. Hay, E. Hevia and D. C. pryde, Tetrahedron Lett., 2011, 52, 4590 CrossRef CAS PubMed.
- Selected publications dealing with the C-4 ortho-lithiation of pyridine derivatives using alkyllithium bases: (a) T. Güngör, F. Marsais and G. Quéguiner, Synthesis, 1982, 499 CrossRef; (b) J. A. Turner, J. Org. Chem., 1982, 48, 3401 CrossRef; (c) M. R. Winkle and R. C. Ronald, J. Org. Chem., 1982, 47, 2101 CrossRef CAS; (d) S. Sengupta and V. Snieckus, Tetrahedron Lett., 1990, 31, 4267 CrossRef CAS; (e) D. L. Comins and M. O. Killpack, J. Org. Chem., 1990, 53, 69 CrossRef; (f) K. Smith, G. A. El-Hiti, M. B. Alshammari and A. Fekri, Synthesis, 2013, 3426 CrossRef CAS PubMed.
- Selected recent reports about the rapid functionalization of oxetanes: (a) D. I. Coppi, A. Salomone, F. M. Perna and V. Capriati, Chem. Commun., 2011, 47, 9918 RSC; (b) J. V. Geden, B. O. Beasley, G. J. Clarkson and M. Shipman, J. Org. Chem., 2013, 78, 12243 CrossRef CAS PubMed; (c) K. F. Morgan, I. A. Hollingsworth and J. A. Bull, Chem. Commun., 2014, 50, 5203 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc03766a |
| This journal is © The Royal Society of Chemistry 2014 |