 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfonyl chlorides as an efficient tool for the postsynthetic modification of Cr-MIL-101-SO3H and CAU-1-NH2†
Arne
Klinkebiel
a,
Nele
Reimer
b,
Martin
Lammert
b,
Norbert
Stock
b and
Ulrich
Lüning
*a
aOtto-Diels-Institut für Organische Chemie, Olshausenstr. 40, 24098 Kiel, Germany. E-mail: luening@oc.uni-kiel.de
bInstitut für Anorganische Chemie, Max-Eyth-Strasse 2, 24118 Kiel, Germany. E-mail: stock@ac.uni-kiel.de
First published on 7th July 2014
Abstract
Postsynthetic modification can be used to introduce sulfonamide functionalities into MOF frameworks. Using sulfonyl chlorides as reactive intermediates, Cr-MIL-SO3H and CAU-1-NH2 have been further modified to give hitherto unknown functionalized MOFs in which a sulfonamide group is bound to the framework either by its N or its S atom.
Among the classes of porous materials, metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) represent a highly versatile group of compounds.1 By varying the nature of the organic linker as well as the coordination environment of the constituting metal ions, these materials offer numerous opportunities to vary physical and chemical properties of the pore surface. They are promising materials for applications such as gas adsorption, separation, catalysis, chemical sensing, drug delivery.2 Variation of the functionality of the organic linker molecules can be achieved by postsynthetic modification.3 The introduction of already functionalized linkers is limited, due to the possible interference of the functional groups in the formation of the desired framework or their incompatibility with the reaction conditions. Nevertheless, numerous presynthetically modified MOFs have been described (e.g. amino,4 hydroxyl,5 formyl,6 alkyne7 and azide8 containing structures). From these functionalized MOFs, postsynthetic modification opens a wide field of different derivatives like amides,4f,9 imines,10 ureas and triazoles.7,8,11 To expand the available synthetic opportunities, it is of great importance to develop new synthetic strategies and methods for modifying functional groups which have not been successfully functionalized further until now. For example, the synthesis of frameworks containing non-coordinating acidic groups is difficult and therefore reports on postsynthetic modifications are rare.12
The class of sulfonamides, which is well-known for its antibacterial effects acting as antimetabolites13 or their catalytical properties,14 can be synthesized from sulfonic acids and amines. The chemical and electronic properties of the sulfonamide group are of great importance. Due to the strong electron-withdrawing effect of the sulfonyl group, the amide proton is acidic. Compared to carbonamides, a sulfonamide is more stable under basic aqueous conditions and the hydrogen-bond strength is enhanced.15
Here we show that the class of sulfonamides is accessible by tandem post-modification of the Cr-MIL-101-SO3H using the corresponding sulfonyl chloride as a reactive intermediate. Extending our concept by treating an amino-containing network like CAU-1-NH24f with sulfonyl chlorides, it is possible to synthesize MOFs with a reversed orientation of the sulfonamide functionality as well.
The MIL-101 framework which is composed of chromium(III), 2-sulfoterephthalate and 2-sulfonate-terephthalate anions reported by Kitagawa and co-workers16 is one of a few existing MOFs17 containing a non-coordinating sulfonic acid. To the best of our knowledge, there has been no postsynthetic modification of this group described until now. We modified it to different N-alkyl and N-aryl substituted sulfonamides (3a–f) as well as an N-pyridine substituted one (3g) (Scheme 1).
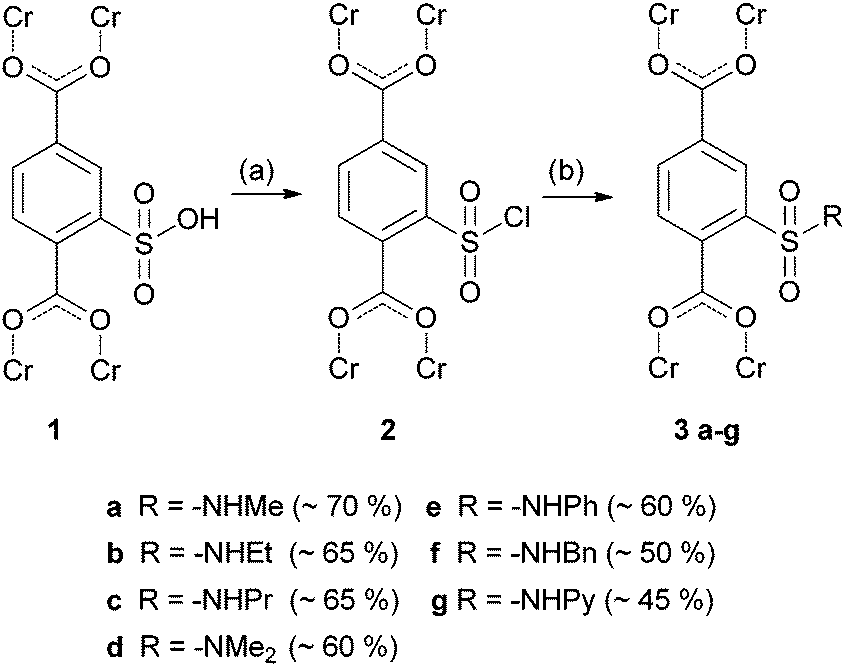 | ||
| Scheme 1 Two-step postsynthetic modification of Cr-MIL-101-SO3H (1) via sulfonyl chloride 2 to produce sulfonamides 3a–g. (a) Oxalyl chloride, cat. DMF, THF, 24 h, room temp. (b) Corresponding amine, THF, 24 h, room temp. | ||
PXRD data (ESI†) demonstrate that all structures are intact after the modification. Infrared spectra of compounds 3a–g (ESI†) show characteristic bands for the introduced alkyl and aryl substituents as well as for sulfonamides. The N2 adsorption isotherms (ESI†) of the functionalized Cr-MIL-101 3a–g samples exhibit the characteristic shape, while the sorption capacity slightly decreases as expected upon postsynthetic modification in comparison to starting material 1. The decrease is only 20% for the methyl substituted sulfonamide 3a, but 50% if benzyl substituted (3f). However for comparison of the different MOFs with one another, the varying degrees of post-synthetic modification (70% for 3a, 50% for 3g) have to be taken into consideration.
For further analysis, the MOFs were dissolved in sodium hydroxide, the insoluble chromium salts were removed, and the remaining solution was analyzed by 1H-NMR data (ESI†). In Fig. 1, spectra for compound 3a are shown as examples. They exhibit the aromatic signals for the desired sulfonamide 3a and the corresponding unfunctionalized sulfoterephthalic acid of the starting material 1. The ratio of the relative integrals allows us to determine the conversion, which is about 70% (Fig. 1(B)). Due to its water solubility, removal of the linker of starting material 1 (Fig. 1(A)) is possible by extracting the functionalized amide of 3a with an organic solvent from an acidic solution, whereas the starting material stays mostly in the aqueous phase.
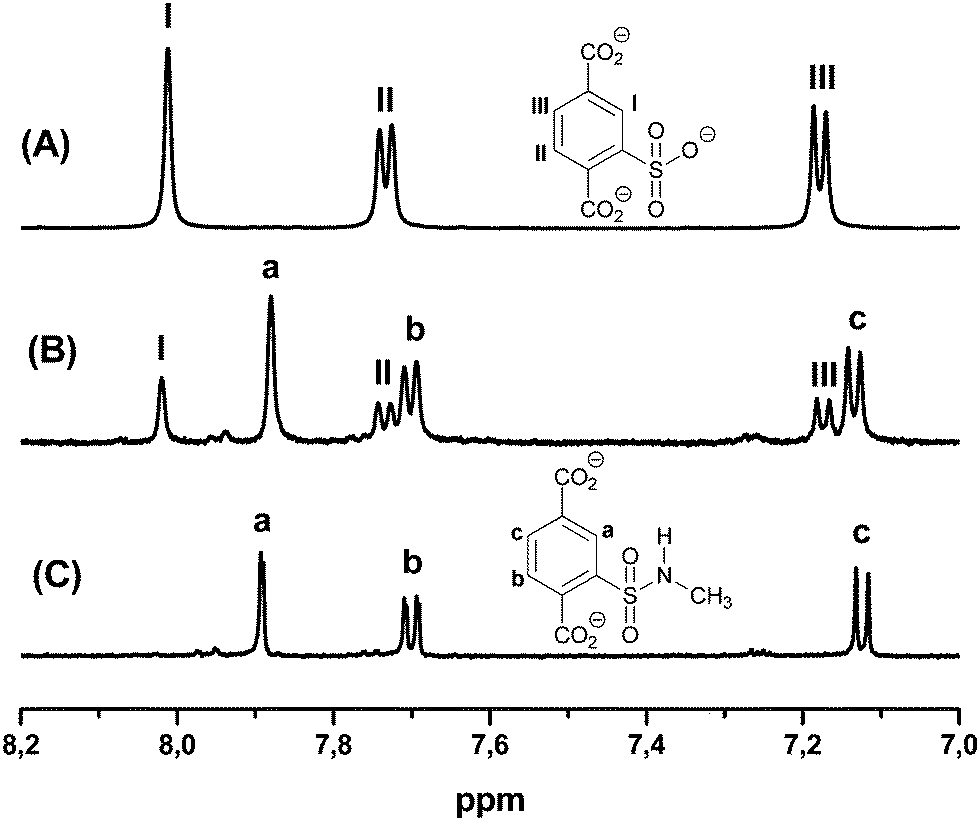 | ||
| Fig. 1 1H-NMR spectra after dissolving the respective MOF in NaOD (aliphatic signals are not shown, see ESI† for details): (A) aromatic protons (I–III) of the unfunctionalized linker of starting material 1, (B) mixture of functionalized amide 3a (a–c) and unfunctionalized linker (I–III) after postsynthetic modification, (C) terephthalic protons (a–c) of the functionalized amide 3a after extraction of the unfunctionalized linker. | ||
Comparison of conversion degrees (Scheme 1) shows a dependency on size of the introduced amine and an upper limit of about 70%. Addition of pyridine or triethylamine to scavenge the generated hydrochloric acid showed no improvement of conversion. Due to the instability of the sulfonyl chloride, the reaction temperature was not increased above 25 °C. The upper limit of conversion matches with the conclusion of Kitagawa16 for one of three sulfonic acid groups to be deprotonated. The resulting charges are compensating the positive charges of the framework.
In all cases, dimethylsulfonamide 3d is formed as a byproduct, which results from the reaction of sulfonyl chloride 2 and dimethylamine, which is formed as a sideproduct from the catalytical amount of dimethylformamide added during the reaction. For verification, we synthesized N,N-dimethylsulfonamide 3d as reference. Variation of the catalytical amount of dimethylformamide from 15 μL to 5 μL showed a significant decrease of this sideproduct, which is shown exemplarily for sulfonamide 3c in Fig. 2. The amount of sideproduct formed varies between 1–5% (see ESI† for details).
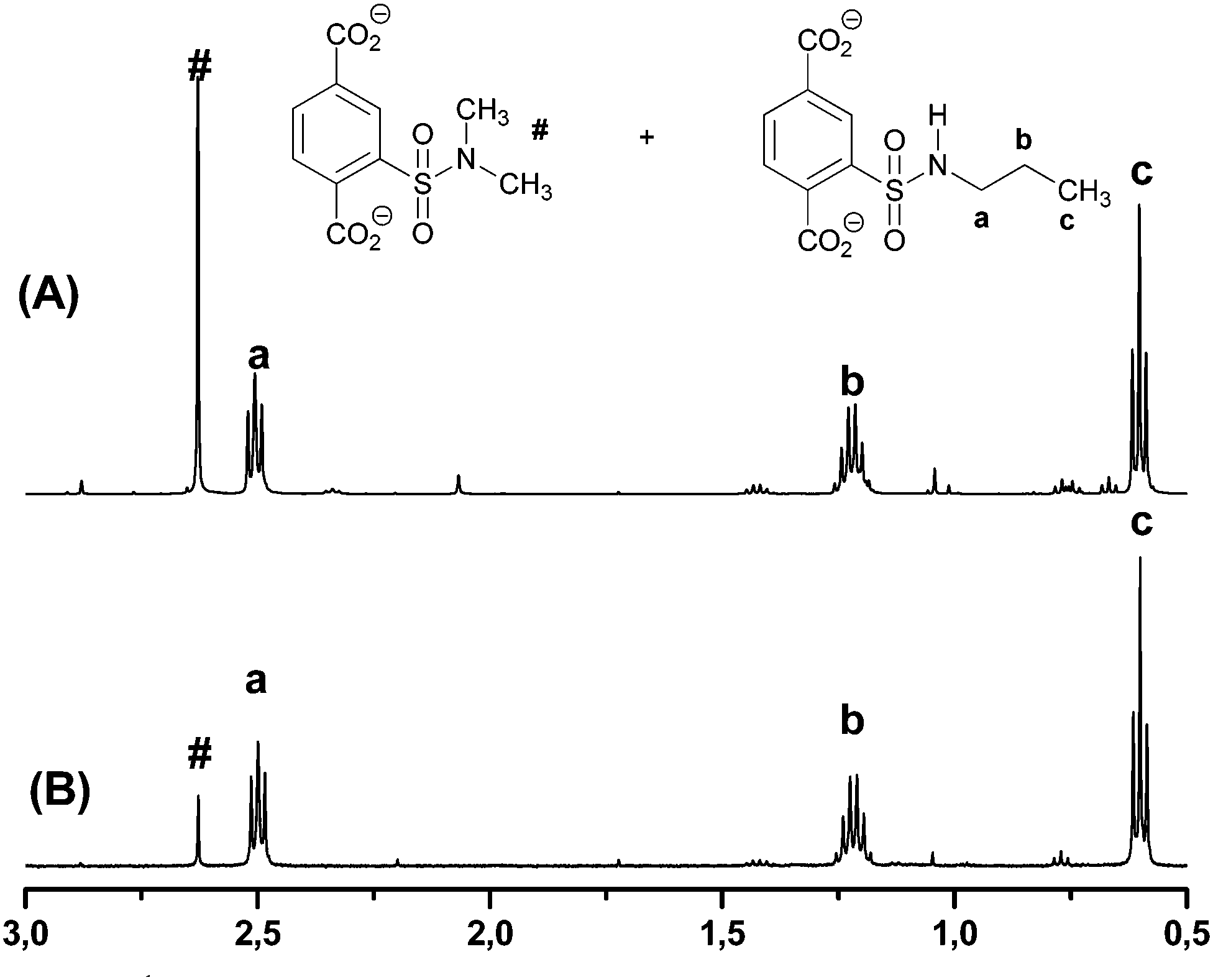 | ||
| Fig. 2 1H-NMR spectra of the respective MOF in NaOD (aromatic signals are not shown, see ESI† for details): N-propyl sulfonamide 3c (a–c) and side product N,N-dimethyl sulfonamide 3d (#) after using (A) 15 μL and (B) 5 μL DMF as catalyst. | ||
To extend our modification concept of using sulfonyl chlorides as intermediates, it was our next goal to invert the binding sequence of the sulfonamide by treating the amino containing framework CAU-1-NH24f (5) with sulfonyl chlorides in order to obtain related amides 4 and 6 (Scheme 2). The synthesis of sulfonamide 6 was carried out at room temperature due to the instability of 2-pyridinylsulfonyl chloride (ESI†), which was synthesized according to a literature procedure.18 To obtain an N-alkyl substituted sulfonamide, we treated compound 5 with methyl sulfonyl chloride, which can withstand higher temperatures (see Table 1).
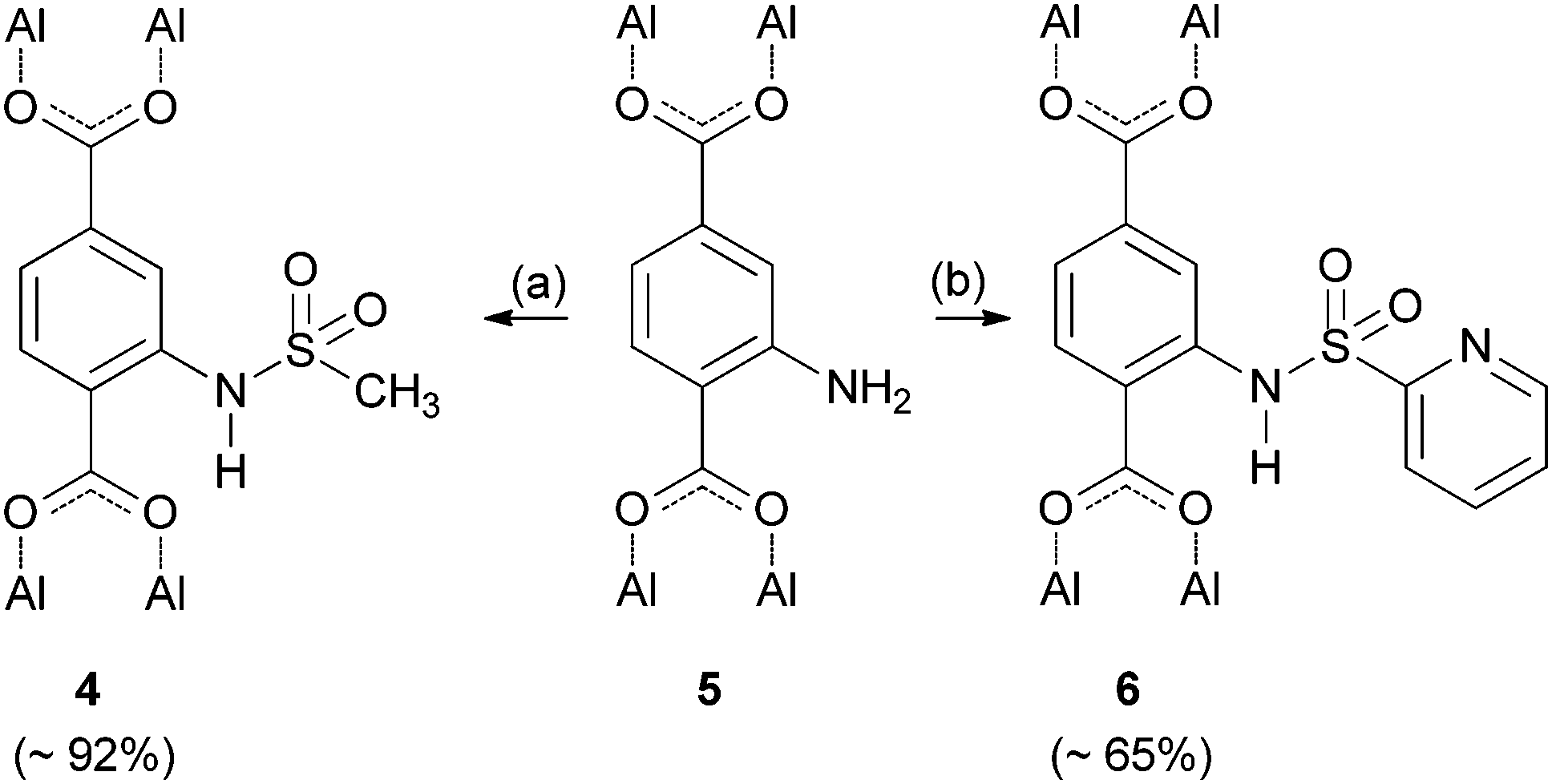 | ||
| Scheme 2 Postsynthetic modification of CAU-1-NH2 (5) using methyl and 2-pyridinylsulfonyl chloride giving the sulfonamides 4 and 6. (a) For different reaction conditions see Table 1. (b) 2-Pyridinylsulfonyl chloride, pyridine, dichloromethane, 24 h, room temp. | ||
| Catalyst | Temperature | Reaction time | Conversion (%) |
|---|---|---|---|
| — | 60 °C | 4 d | 59 |
| Pyridine | 60 °C | 24 h | 92 |
| — | 90 °C (mw) | 30 min | 43 |
| — | 120 °C (mw) | 30 min | 64 |
| — | 120 °C (mw) | 60 min | 76 |
| — | 150 °C (mw) | 30 min | 82 |
| — | 180 °C (mw) | 30 min | 91 |
| Pyridine | 110 °C (mw) | 10 min | 92 |
The ratio of the relative integrals of the desired sulfonamides and the corresponding amino terephthalic acid 5 in the 1H-NMR spectra of the dissolved material (ESI†) allows an approximation of the conversion of about 65% for pyridinesulfonamide 6 and a conversion of 92% to methylsulfonamide 4. In contrast to the previous approaches using Cr-MIL-101-SO3H, the addition of pyridine as a catalyst and scavenger for the generated hydrochloric acid showed a significant improvement of conversion in both cases.
Due to the stability of methylsulfonyl chloride at higher temperatures, the reaction conditions for the synthesis of sulfonamide 4 were optimized by performing the reaction in a normal glass vessel as well as under microwave conditions.
In summary, we report a synthetic strategy for introducing the sulfonamide functionality into MOFs for the first time. Amino and sulfonic acid containing MOFs like CAU-1-NH2 and Cr-MIL-101-SO3H can be modified by using sulfonyl chlorides as reactive intermediates in two different routes. With the broad variation of substituents and the different orientations of the sulfonamide moiety to the framework, this method offers a promising tool for the postsynthetic modification of amino and sulfonic acid containing frameworks. Above all, the sulfonamide group is a powerful functionality with respect to catalysis and host–guest interactions. Their implementation into MOFs makes them promising materials for future developments.
Financial support of the Deutsche Forschungsgemeinschaft (SPP 1362) is gratefully acknowledged.
Notes and references
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 ( Angew. Chem. , 2004 , 116 , 2388 ) CrossRef CAS PubMed; (c) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (d) D. Farusseng, Metal-Organic Frameworks, Wiley-VCH, Weinheim, 2011 Search PubMed.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, (ed.) Chem. Rev., 2012, 112, 673; (b) J. R. Long and O. M. Yaghi, (ed.) Chem. Soc. Rev., 2009, 38, 1201; (c) C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed; (d) S. H. Jhung, N. A. Khan and Z. Hasan, CrystEngComm, 2012, 14, 7099 RSC; (e) S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249 CrossRef CAS PubMed; (f) B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014 10.1039/c4cs00006d; (g) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014 10.1039/c3cs60442j; (h) E. Coronado and G. Mínguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- (a) S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568 CrossRef CAS PubMed; (b) J. S. Costa, P. Gamez, C. A. Black, O. Roubeau, S. J. Teat and J. Reedijk, Eur. J. Inorg. Chem., 2008, 1551 CrossRef CAS; (c) K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677 ( Angew. Chem. , 2008 , 120 , 689 ) CrossRef CAS PubMed; (d) Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296 CrossRef CAS PubMed; (e) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed; (f) T. Ahnfeldt, N. Guillou, D. Gunzelmann, I. Margiolaki, T. Loiseau, G. Férey, J. Senker and N. Stock, Angew. Chem., Int. Ed., 2009, 48, 5163 ( Angew. Chem. , 2009 , 121 , 5265 ) CrossRef CAS PubMed.
- T. Ahnfeldt and N. Stock, CrystEngComm, 2012, 14, 505 RSC.
- W. Morris, C. J. Doonan, H. Furukawa, R. Banerjee and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 12626 CrossRef CAS PubMed.
- T. Gadzikwa, G. Lu, C. L. Stern, S. R. Wilson, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 5493 RSC.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354 CrossRef CAS PubMed.
- (a) S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341 CrossRef CAS PubMed; (b) D. Britt, C. Lee, F. J. Uribe-Romo, H. Furukawa and O. M. Yaghi, Inorg. Chem., 2010, 49, 6387 CrossRef CAS PubMed.
- (a) M. J. Ingleson, J. P. Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 2680 RSC; (b) C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492 CrossRef CAS PubMed.
- (a) T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48, 3057 CrossRef CAS PubMed; (b) M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518 CrossRef CAS PubMed.
- (a) N. Reimer, B. Gil, B. Marszalek and N. Stock, CrystEngComm, 2012, 14, 4119 RSC; (b) M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304 CrossRef CAS PubMed.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, 1995 Search PubMed.
- (a) T. Kano, R. Sakamoto, Y. Yamaguchi, K. Itoh and K. Maruoka, Chem. Commun., 2013, 49, 1118 RSC; (b) L. Zu, H. Xie, H. Li, J. Wang and W. Wang, Org. Lett., 2008, 10, 1211 CrossRef CAS PubMed; (c) Z. Xu, R. Wang, J. Xu, C.-S. Da, W.-J. Yan and C. Chen, Angew. Chem., Int. Ed., 2003, 42, 5747 ( Angew. Chem. , 2003 , 115 , 5925 ) CrossRef CAS PubMed.
- C. S. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 ( Angew. Chem. , 2004 , 116 , 5424 ) CrossRef CAS PubMed.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294 CrossRef CAS PubMed.
- (a) S. Biswas, J. Zhang, Z. Li, Y.-Y. Liu, M. Grzywa, L. Sun, D. Volkmer and P. Van Der Voort, Dalton Trans., 2013, 42, 4730 RSC; (b) M. Lin Foo, S. Horike, T. Fukushima, Y. Hijikata, Y. Kubota, M. Takata and S. Kitagawa, Dalton Trans., 2012, 41, 13791 RSC; (c) Postsynthetic partial sulfatation: M. G. Goesten, J. Juan-Alcañiz, E. V. Ramos-Fernandez, K. B. S. S. Gupta, E. Stavitski, H. van Bekkum, J. Gascon and F. Kapteijn, J. Catal., 2011, 281, 177 CrossRef CAS PubMed.
- M. Kajino, A. Hasuoka and H. Nishida, US Pat., US 20070060623A1, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H-NMR spectroscopy, FT-IR spectroscopy, XRPD data, adsorption isotherms. See DOI: 10.1039/c4cc03746d |
| This journal is © The Royal Society of Chemistry 2014 |