 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalysed carboxylation of organoboronates†
Yusuke
Makida
a,
Enrico
Marelli
a,
Alexandra M. Z.
Slawin
a and
Steven P.
Nolan
*ab
aEaStCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: snolan@st-andrews.ac.uk
bChemistry Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
First published on 5th June 2014
Abstract
A nickel/N-heterocyclic carbene (NHC) catalysed carboxylation of aryl-, heteroaryl- and alkenylboronates, affording the corresponding carboxylic acids, has been developed. This transformation proceeds under one atmosphere of CO2 with a broad range of substrates and exhibits good functional group compatibility.
Carbon dioxide (CO2) is one of the most promising carbon sources for the production of fine chemicals as it is abundant, non-toxic, cheap and renewable.1 Amongst the numerous reports regarding CO2 fixation mediated by metal centres, significant efforts have targeted the development of efficient and direct carboxylation protocols.2 Highly reactive organometallic reagents, such as organolithium and Grignard reagents, are the historical, yet still typical starting materials for nucleophilic carboxylation, since they are reactive enough to overcome the inherent thermodynamic stability of CO2. However, their narrow functional group compatibility hampers their general and practical use. Consequently, carboxylation using a widely adaptable reagent is highly desirable.3–9 In the last decade, the transition metal catalysed carboxylation of organoboronates with CO2 has been achieved using Rh,3 Cu4 and Ag5 catalysts (Scheme 1, path A).
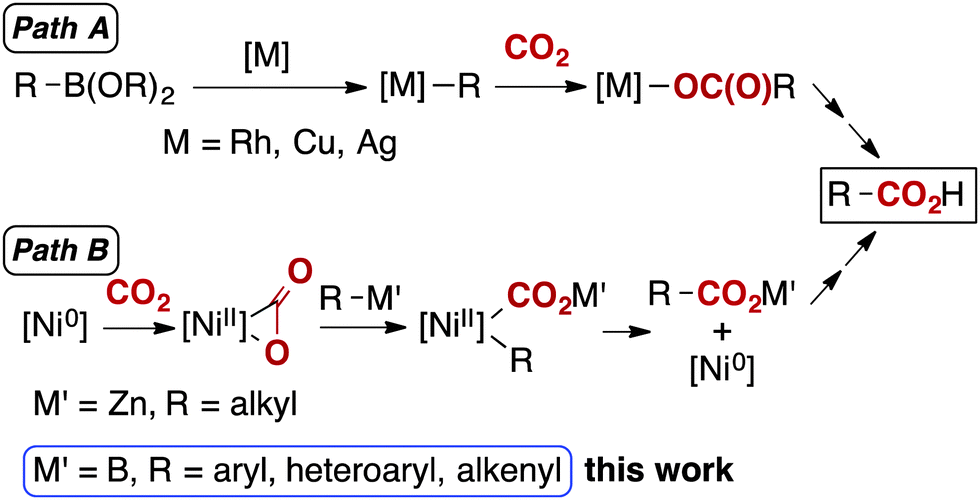 | ||
| Scheme 1 Two distinct pathways for the metal-catalysed carboxylation of less-nucleophilic organometallic reagents. | ||
Yorimitsu, Oshima and Dong have described a nickel-catalysed carboxylation employing organozinc reagents with CO2, and proposed that an Aresta-type complex was involved in the catalytic cycle (Scheme 1, path B, M′ = Zn).7,10 While these reactions were restricted mostly to alkylzinc reagents,11 nickel-based systems offer fascinating possibilities in terms of adopting a mechanism distinct from those reported with other transition metals (Scheme 1). Nickel catalysis typically proceeds through oxidative cyclization of CO2 by zero-valent nickel, followed by a transmetalation and reductive elimination (Scheme 1, path B). Other transition metal catalysts normally involve transmetalation followed by insertion of CO2 into a metal–carbon bond without a change in metal oxidation state (Scheme 1, path A).
Herein, we report a methodology for the carboxylation of organoboronates under one atmosphere of CO2 catalysed by a well-defined nickel pre-catalyst having a specific N-heterocyclic carbene ligand (Scheme 1, path B, M′ = B). This reaction presents broad functional group compatibility and simple reaction conditions. Moreover, to the best of our knowledge, the nickel-catalysed coupling of organoboron reagents with CO2 has not been reported to date.
Very recently, we reported the synthesis and characterization of novel [Ni(NHC)(allyl)Cl] complexes, bearing the flexible yet bulky NHC ligands IPr* and IPr*OMe; these complexes are useful catalysts for C–N and C–S coupling reactions.12 Although they generate Ni(0) active species easily with the assistance of the steric pressure of the bulky NHC ligand and the labile η3-allyl moiety, these complexes possess good stability. In this context, we began to explore the possibility of an efficient carboxylation methodology using this catalytic nickel system in conjunction with organoboron reagents and CO2 as the reaction partners.
We began our survey by screening the reactivity of nickel precatalysts 1a–g, as depicted in Table 1. [Ni(NHC)(allyl)Cl] complexes bearing less sterically demanding NHCs (IPr, SIPr, IMes and SIMes, see Fig. 1 for key structural features of NHC used in this study) showed poor to moderate reactivities (entries 1–4). For example, the reaction of boronate 2a using [Ni(IPr)(allyl)Cl] as precatalyst gave the carboxylation product 3a in only 7% NMR conversion.13 The catalytic activity was drastically shifted by a judicious selection of the NHC ligand. Indeed, the reactions using the bulkier IPr*-based ligands (IPr* and IPr*OMe) afforded the corresponding carboxylic acids quantitatively (entries 5 and 9). At 80 °C, the IPr* ligand showed higher reactivity than IPr*OMe (entries 6 and 10). The effect of the steric hindrance may very well result in an improvement of the stability of the active species or in the acceleration of one of the catalytic steps. Other observations concerning the optimized reaction conditions are to be noted: a 5 mol% catalyst loading is required for excellent conversion (entry 5 vs. 7); the analogous nickel complex with a Cp moiety, instead of an allyl group, showed no activity (entry 8);12a a catalytic species prepared in situ from [Ni(cod)2] and IPr* led to high conversion (entry 11);14 ethers could be used but polar or chlorinated solvents gave no conversion (entries 12–15); despite its ability to activate boron reagents, CsF displays no reactivity as a base, (entry 16).3,4a The use of hydroxides and carbonates also gave no conversion (entries 19 and 20). When a sub-stoichiometric amount of KOtBu was used, the product was formed sub-stoichiometrically (entry 17) and carboxylation did not proceed in the absence of the nickel pre-catalyst (entry 18).
|
|
||||
|---|---|---|---|---|
| Entry | Ni pre-catalyst | T (°C) | Solvent | y (%) |
| a Reaction conditions: 2a (0.2 mmol, 1.0 equiv.), CO2 (1 atm), Ni precatalyst (5 mol%), KOtBu (0.24 mmol), solvent (1.0 mL), 15 h. b Yield determined by 1H NMR spectroscopic analysis of crude product after acidic work up using 1,2-diphenylethane as an internal standard; average of two runs. c With 1.0 mol% of 1e. d With 5.0 mol% of [Ni(cod)2] and 6.0 mol% of IPr*. e With 0.24 mmol of CsF instead of KOtBu. f With 0.03 mmol of KOtBu. g Without Ni precatalyst. h 0.24 mmol of KOH used instead of KOtBu. i 0.24 mmol of K2CO3 used instead of KOtBu. | ||||
| 1 | [Ni(IPr)(all)Cl] (1a) | 100 | Toluene | 7 |
| 2 | [Ni(SIPr)(all)Cl] (1b) | 100 | Toluene | 45 |
| 3 | [Ni(IMes)(all)Cl] (1c) | 100 | Toluene | 15 |
| 4 | [Ni(SIMes)(all)Cl] (1d) | 100 | Toluene | 10 |
| 5 | [Ni(IPr*)(all)Cl] (1e) | 100 | Toluene | Quant. |
| 6 | 1e | 80 | Toluene | 56 |
| 7c | 1e | 100 | Toluene | 15 |
| 8 | [Ni(IPr*)(Cp)Cl] (1f) | 100 | Toluene | 0 |
| 9 | [Ni(IPr*OMe)(all)Cl] (1g) | 100 | Toluene | Quant. |
| 10 | 1g | 80 | Toluene | 26 |
| 11d | [Ni(cod)2]/IPr* | 100 | Toluene | Quant. |
| 12 | 1e | 80 | Dioxane | 37 |
| 13 | 1e | 80 | CPME | 47 |
| 14 | 1e | 80 | DMA | 0 |
| 15 | 1e | 80 | DCE | 0 |
| 16e | 1e | 100 | Toluene | 0 |
| 17f | 1e | 100 | Toluene | 10 |
| 18g | — | 100 | Toluene | 0 |
| 19h | 1e | 100 | Toluene | 0 |
| 20i | 1e | 100 | Toluene | 0 |

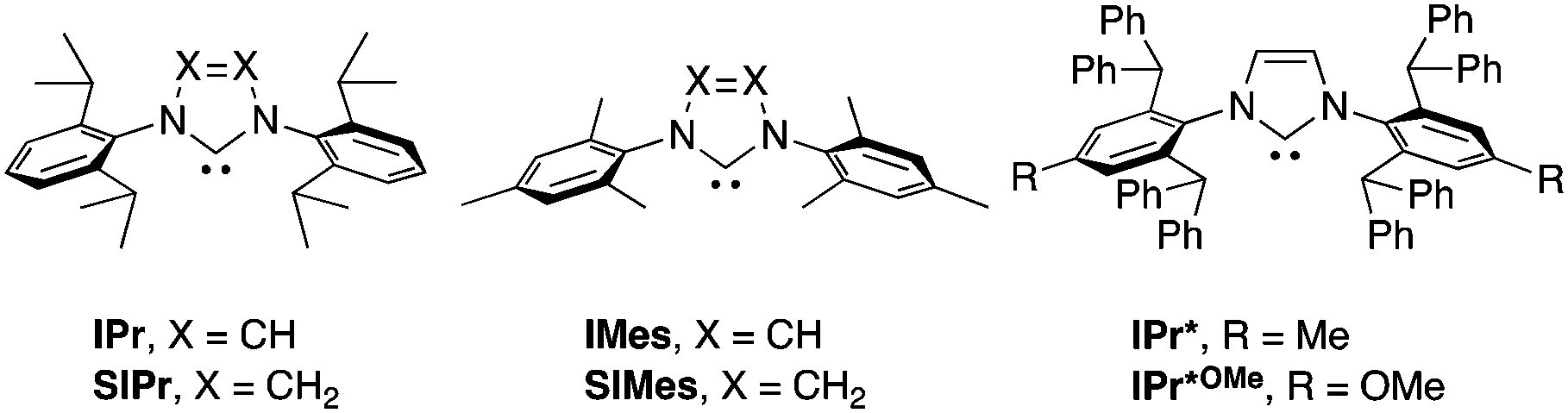 | ||
| Fig. 1 Structures of NHC ligands used in this study. | ||
Importantly, possible side-reactions such as homocoupling or protodeborylation were not observed and the starting boronate was the only recovered material when reactions did not proceed efficiently to product. Other arylboron reagents such as boronic acids, pinacolates, and potassium trifluoroborates were tested but resulted in almost no reaction.13,15 Thus, conditions employed in entry 5 were selected as the optimum reaction settings.
We next evaluated the carboxylation of a range of aryl- and heteroarylboronates with CO2 under these optimized conditions. As shown in Table 2, p-anisic acid (3a), benzoic acid (3b) and p-toluic acid (3c) were obtained in excellent yields (entries 1–3). In addition, this nickel-catalysed carboxylation tolerated a variety of functional groups at the para position of the arylboronate reagent: examples include silyl ether, CF3 and tertiary amino groups, as well as halides (entries 4–8).
|
|
||||
|---|---|---|---|---|
| Entry | Product | Yieldb (%) | ||
| a Reaction conditions: 2 (0.4 mmol), CO2 (1 atm), 1e (5 mol%), and KOtBu (0.48 mmol) in toluene (2.0 mL) at 100 °C for 15 hours. b Yield of isolated product; average of two runs. c 10 mol% 1e. | ||||
| 1 |
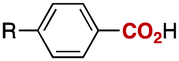
|
R = | OMe (3a) | 96 |
| 2 | H (3b) | 99 | ||
| 3 | Me (3c) | 99 | ||
| 4 | OTBDMS (3d) | 90 | ||
| 5 | NMe2 (3e) | 84 | ||
| 6 | F (3f) | 96 | ||
| 7 | Cl (3g) | 83 | ||
| 8 | CF3 (3h) | 97 | ||
| 9c | CO2Me (3i) | 90 | ||
| 10 | CN (3j) | 97 | ||
| 11 |
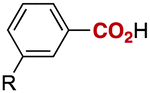
|
R = | OMe (3k) | 96 |
| 12 | F (3l) | 78 | ||
| 13 | CF3 (3m) | 96 | ||
| 14 | CO2Me (3n) | 90 | ||
| 15 |
|
R = | OMe (3o) | 92 |
| 16 | F (3p) | 81 | ||
| 17c |
|
3q | 88 | |
| 18 |

|
3r | 93 | |
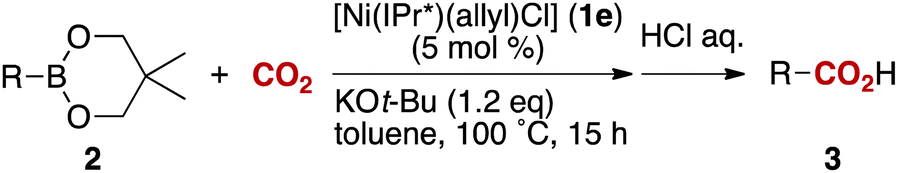
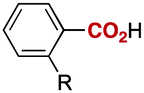
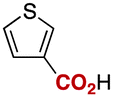
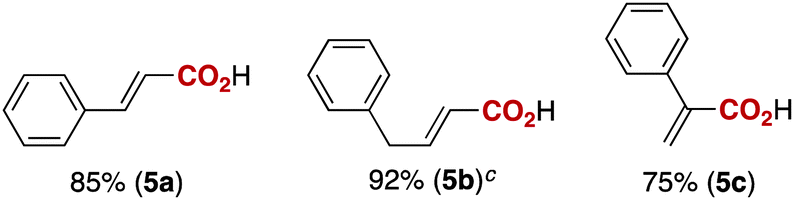
Notably, electrophilic and base-sensitive functionalities were also compatible, giving the corresponding benzoic acid derivatives 3i (90%) and 3j (97%), respectively, while leaving the ester and nitrile moieties untouched (entries 9 and 10). This transformation was also applicable to both electron-rich and electron-deficient substituents in the meta position (entries 11–14). The current limitation of the method lies with the poor tolerance of ortho-substituted arylboronates. For example, the reaction of the neopentylgycolate of o-tolylboronic acid resulted in no reaction. Although the reaction proved to be sensitive to proximal steric bulk, o-anisic acid (3o) and 2-fluorobenzoic acid (3p) could be synthesised (92% and 81% respectively) (entries 15 and 16). Heteroaromatic boronate 2q reacted with CO2 to provide 3-thenoic acid (3q) in 88% yield with 10 mol% catalyst (entry 17). Boronate 2r was more reactive, affording 2-furoic acid (3r) in 93% yield (entry 18). The protocol was successfully extended to alkenylboron reagents (Table 3).16 Boronates having a conjugated π-system 4a and non-conjugated 4b reacted efficiently to afford the corresponding α,β-unsaturated carboxylic acids 5a and 5b in 85% and 92% yield, respectively. exo-Olefinic boronate 4c also showed good reactivity (75%).
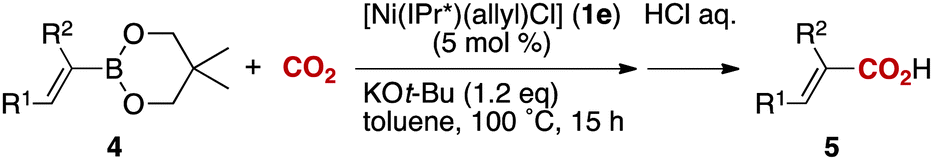
As there is such a profound influence of the boron reagents, initial studies focused on the alkoxide base and how it could modify the boron reagent. It was originally thought that the role of KOtBu was to activate the boronates for transmetalation. Indeed, the reaction between 2 and KOtBu gave the corresponding aryltrialkoxyborate 6, quantitatively (eqn (1), see ESI† for more details). This boron quaternization proceeds smoothly at room temperature and reached completion within 30 minutes as monitored by NMR spectroscopy.13,17 The atom connectivity in 6f was unambiguously confirmed by X-ray analysis on single crystals (Fig. 2).
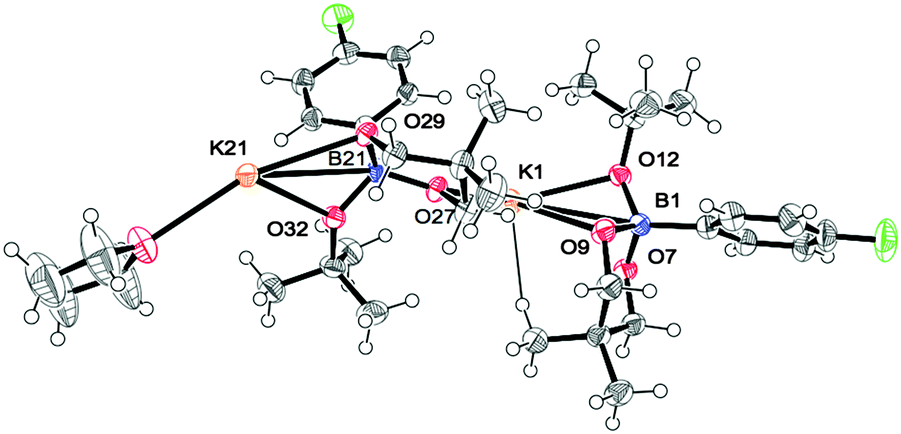 | ||
| Fig. 2 Molecular structure of 6f: (6f)2·THF. | ||
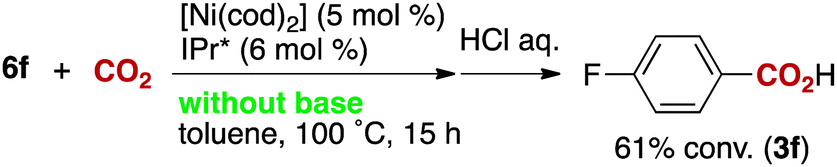
The reactivity of the borate 6f with CO2 under nickel catalysis was then examined. Isolated 6f was treated with CO2 in the presence of 5 mol% of 1e and 7 mol% of KOtBu (eqn (2)), and the reaction afforded >95% conversion to 3f. In addition, the reaction proceeds with slightly lower reactivity in the absence of KOtBu (eqn (3)). If a nickel(0) precursor is used, entirely base-free conditions can be achieved (61% conversion, eqn (4)).
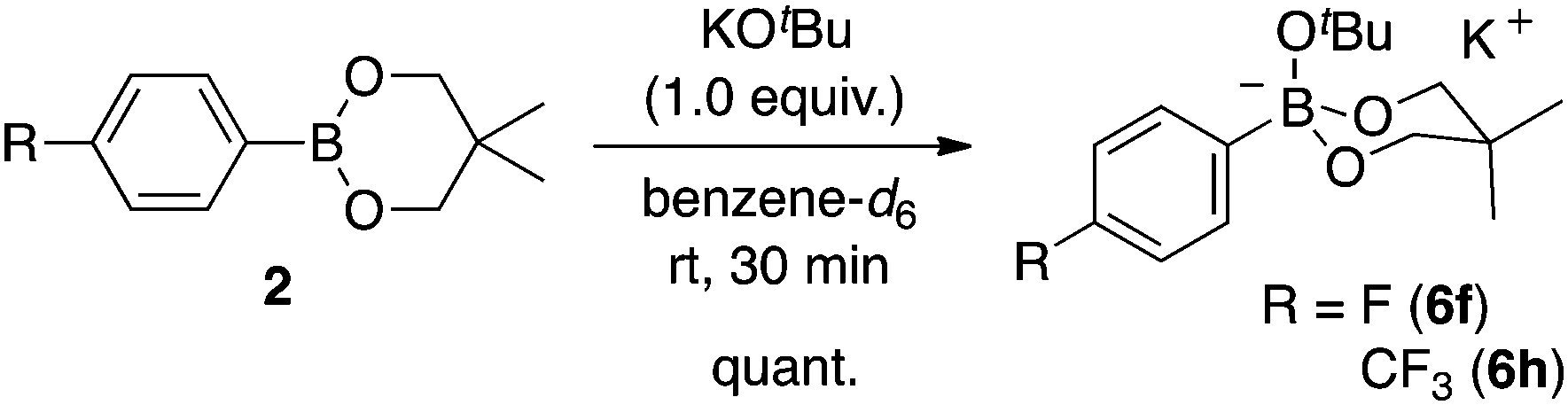 | (1) |
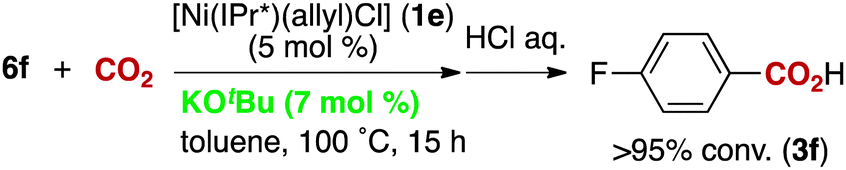 | (2) |
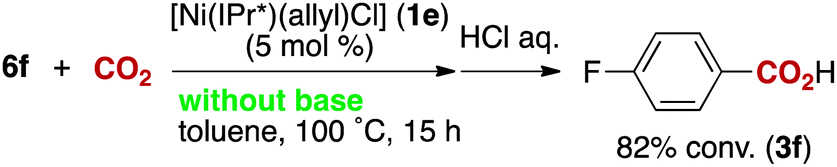 | (3) |
 | (4) |
These results support our hypothesis that organoborates 6 are the reactive species in this reaction.
In summary, we have developed a novel Ni-catalysed strategy for CO2 incorporation using organoboron reagents. IPr*-based ligands show exceptional reactivity for this transformation. Substituted boronates with various functionalities are easily accessible and can be coupled with CO2 using nickel catalysis. The substrate scope is complementary to the reaction of organozinc reagents, which is mainly suitable for alkylzinc reagents. Ongoing mechanistic and catalytic studies are aimed at understanding the critical role of the ligand in this and related catalytic transformations.
We thank the ERC (Advanced Researcher award-FUNCAT, CO2Chem) and King Saud University for funding and the EPSRC NMSSC in Swansea for mass spectrometric analyses. SPN is a Royal Society Wolfson Research Merit Award holder.
Notes and references
- For reviews regarding CO2 fixation, see: (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (b) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (c) Carbondioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed; (d) M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC; (e) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS; (f) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2013, 114, 1709–1742 CrossRef PubMed; (g) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- For reviews on metal-catalysed carboxylation, see: (a) A. Correa and R. Martín, Angew. Chem., Int. Ed., 2009, 48, 6201–6204 CrossRef CAS PubMed; (b) R. Martín and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef PubMed; (c) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (d) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (e) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC; (f) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC; (g) X. Cai and B. Xie, Synthesis, 2013, 3305–3324 CrossRef CAS PubMed.
- K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706–8707 CrossRef CAS PubMed.
- (a) J. Takaya, S. Tadami, K. Ukai and N. Iwasawa, Org. Lett., 2008, 10, 2697–2700 CrossRef CAS PubMed; (b) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef CAS PubMed; (c) P. J. Riss, S. Lu, S. Telu, F. I. Aigbirhio and V. W. Pike, Angew. Chem., Int. Ed., 2012, 51, 2698–2702 CrossRef CAS PubMed; (d) W. Wang, G. Zhang, R. Lang, C. Xia and F. Li, Green Chem., 2013, 15, 635–640 RSC.
- X. Zhang, W.-Z. Zhang, L.-L. Shi, C.-X. Guo, L.-L. Zhang and X.-B. Lu, Chem. Commun., 2012, 48, 6292–6294 RSC.
- For Pd-catalysed carboxylation of allylstannanes, see: (a) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057–5058 CrossRef CAS; (b) R. Johansson and O. F. Wendt, Dalton Trans., 2007, 488–492 RSC; (c) J. Wu and N. Hazari, Chem. Commun., 2011, 47, 1069–1071 RSC.
- For Ni- and Pd-catalysed carboxylation of organozinc reagents, see: (a) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681–2683 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed.
- For Cu-catalysed carboxylation of alkylboranes, see: (a) H. Ohmiya, M. Tanabe and M. Sawamura, Org. Lett., 2011, 13, 1086–1088 CrossRef CAS PubMed; (b) T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114–8117 CrossRef CAS PubMed.
- For other selected Ni-catalysed carboxylation processes, see: (a) M. Takimoto and M. Mori, J. Am. Chem. Soc., 2002, 124, 10008–10009 CrossRef CAS PubMed; (b) C. M. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936–14937 CrossRef CAS PubMed; (c) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106–9109 CrossRef CAS PubMed; (d) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221–1224 CrossRef PubMed; (e) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062–1069 CrossRef CAS PubMed.
- M. Aresta, F. C. Nobile, V. G. Albano, E. Forni and M. Manassero, Chem. Commun., 1975, 636–637 RSC.
- Only phenylzinc as an arylzinc reagent could be used, as an exception. See ref. 7.
- (a) A. R. Martin, D. J. Nelson, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 3127–3313 CrossRef CAS; (b) A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265–6270 CrossRef CAS.
- See ESI† for experimental details.
- Other Ni(0) precursors were also tested: for example, using [Ni(SIPr)(η6-toluene)] as catalyst resulted in a carboxylic acid yield of 32%; see: Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Organometallics, 2014, 33, 1276–1282 CrossRef CAS.
- When pinacolboranes were used, trace amounts of homocoupling products were detected by 1H NMR spectroscopy.
- This class of products can be obtained by hydrocarboxylation of alkynes: see: T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523–527 CrossRef CAS PubMed and references therein.
- Similar activation pathways have been reported: (a) B. Saito and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 9602–9603 CrossRef CAS PubMed; (b) Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS PubMed; (c) K. L. Billingsley and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 4695–4698 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for all new compounds and all products. CCDC 998382. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03650f |
| This journal is © The Royal Society of Chemistry 2014 |