 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The first structural and spectroscopic characterisation of a ring-opened form of a 2H-naphtho[1,2-b]pyran: a novel photomerocyanine†
Stuart
Aiken
a,
Kathryn
Booth
a,
Christopher D.
Gabbutt
a,
B.
Mark Heron
*a,
Craig R.
Rice
a,
Azzam
Charaf-Eddin
b and
Denis
Jacquemin
bc
aDepartment of Chemical Sciences, School of Applied Science, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: m.heron@hud.ac.uk
bCEISAM, UMR CNRS 6230, Chimie Et Interdisciplinarité, Synthèse, Analyse, Modélisation, Université de Nantes, Faculté des Sciences et des Techniques, BP 92208, 2, rue de la Houssinière, F-44322 Nantes Cedex 3, France
cInstitut Universitaire de France (IUF), 103 blvd St Michel, F-75005 Paris Cedex 5, France
First published on 5th June 2014
Abstract
Heating 4-methoxy-1-naphthol with a 1,1-diarylprop-2-yn-1-ol gave the 2,2-diaryl-6-methoxy-2H-naphtho[1,2-b]pyran together with the novel merocyanine, (E)-2-[3′,3′-bis(aryl)allylidene]-4-methoxynaphthalen-1(2H)-one. Brief UV-irradiation of the pyran favoured the formation of the (Z)-merocyanine with longer irradiation and/or acidic conditions favouring the (E)-isomer.
Naphthopyrans (benzochromenes) and fused carbocyclic and heterocyclic derivatives have attracted significant academic interest as a consequence of their photochromism which has been widely exploited in commercial photochromic ophthalmic sun-lenses.1,2 The photochromic properties of 3H-naphtho[2,1-b]pyrans 1 and 2H-naptho[1,2-b]pyrans 2 (Fig. 1) have been studied and differences in their performance characteristics contrasted.3 Multi-nuclear NMR spectroscopy has been demonstrated to be a useful tool for probing photochromism4 and the structure of the transient merocyanine dyes resulting from the photochemically-induced electrocyclic ring-opening of 3,3-bis(4-fluorophenyl)-3H-naphtho[2,1-b]pyran have been identified as the (Z)- and (E)-1-[3,3-di(4-fluorophenyl)-allylidene]naphthalen-2(1H)-ones 3 and 4, respectively;5 the colourless allenyl naphthol 5 (Ar = 4-FC6H4–) was also identified in a subsequent study.6
 | ||
| Fig. 1 Isomeric naphthopyrans and their photomerocyanine dyes. | ||
Given the commercial interest in 2H-naphtho[1,2-b]pyran derived systems2 it is somewhat remarkable that there have been, to the best of our knowledge, no examples of the characterisation of the proposed merocyanine dyes (6 and 7) resulting from the photochemical ring-opening of the 2H-naphtho[1,2-b]pyran system 2. Furthermore, there does not appear to have been any reports of the differing absorption properties of the isomeric merocyanine dyes. In this communication we report our findings concerning the isolation, characterisation, structure and interconversion of photomerocyanines derived from 6-methoxy-2,2-bis(4-methoxyphenyl)-2H-naphtho[1,2-b]pyran.
Heating a mixture of 4-methoxy-1-naphthol, propynol 8a, PPTS and trimethyl orthoformate in 1,2-dichloroethane under reflux7 gave a multicomponent reaction mixture, which upon elution from silica with 20% EtOAc in hexane, gave 9 (37%), a 2,2-diaryl analogue of the Brazilian hardwood (Paratecoma alba) extractive lapachenole,8 and the more polar, intensely coloured merocyanine 10 (12%) (Scheme 1). We have previously noted the formation of permanent merocyanine dyes, 4-(3,3-diarylylallylidene)naphthalen-1(4H)-ones, as by-products from the reaction between 1-naphthol and 1,1-diarylprop-2-yn-1-ols9 and Coelho et al., have reported the formation of a related dye, 4-(3,3-diphenylallylidene)-8-hydroxynaphthalen-1(4H)-one, derived from 1,8-dihydroxynaphthalene and 1,1-diphenylprop-2-yn-1-ol.10
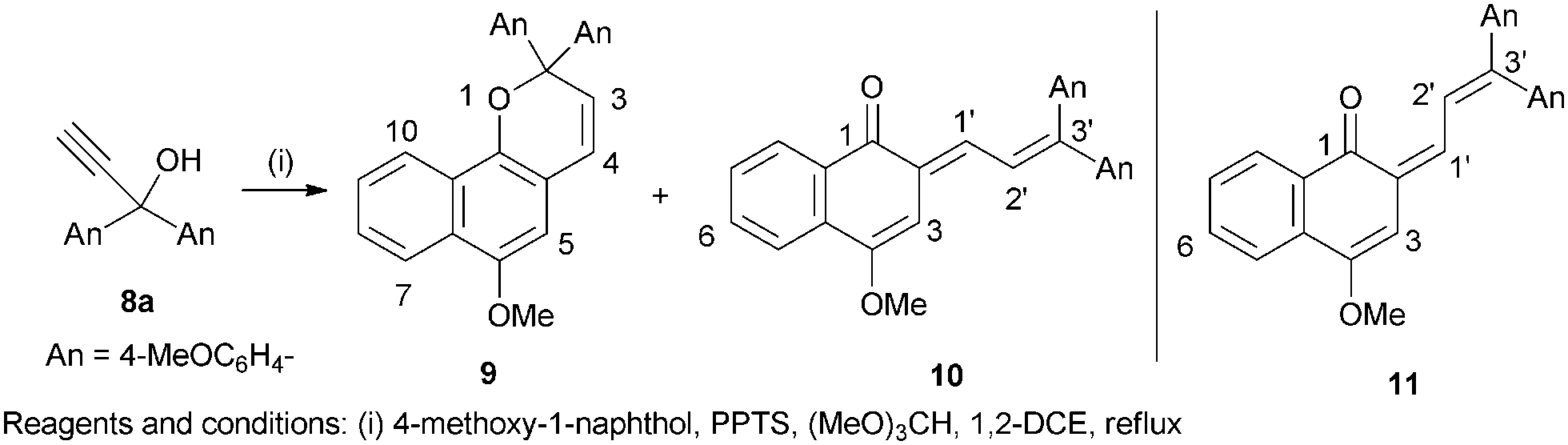 | ||
| Scheme 1 Naphthopyran 9 and merocyanine dyes 10 and 11. | ||
The 1H NMR spectra of 9 (ESI,† Fig. S8) and 10 when recorded in CDCl3 solution after 1 h were complex. From these spectra it was apparent that 9 underwent ring-opening to afford a mixture of 9 and 10, together with a minor amount of an alternate merocyanine proposed as 11 (vide infra). Whilst the 1H NMR spectrum of 10 in CDCl3 solution indicated that ring-closure had occurred to afford the same equilibrium mixture of 9, 10 and 11. Our initial thoughts to account for this behaviour were that 9 and 10 were undergoing photochromic conversion mediated by ambient laboratory daylight resulting in an equilibrium mixture of the three species. However, when freshly preparing the 1H NMR sample of 9 in only low levels of (ambient) laboratory lighting the same instantaneous development of the maroon colour attributed to the ring-opened species 10 (and 11) was noted leading us to consider that it was perhaps residual acidity in the CDCl311 which was the main contributor to the colour development. To explore this postulate a sample of commercial CDCl3 was washed with aqueous K2CO3 followed by water and dried (anhyd. K2CO3)12 and then used to prepare a solution of 9 under low levels of (ambient) laboratory lighting. The resulting solution was paler in colour and the 1H NMR spectrum of 9 recorded after 1 h was quite different from the sample recorded in commercial CDCl3 and displayed signals attributed to a ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of compounds 9:11 (see Fig. 2 and ESI,† Fig. S10), with no evidence of 10; a feature which implies that acidity was important for the isomerisation of 11 to 10.
:1 mixture of compounds 9:11 (see Fig. 2 and ESI,† Fig. S10), with no evidence of 10; a feature which implies that acidity was important for the isomerisation of 11 to 10.
 | ||
| Fig. 2 1H NMR spectrum of 9 in commercial CDCl3 (lower) and base washed CDCl3 (upper) with expansion δ 5.7 → 8.7. | ||
The 1H NMR spectra of freshly prepared d6-acetone solutions (without any protection from (ambient) laboratory daylight) of 9 and 10 were next examined (ESI,† Fig. S4 and S13). Whilst the d6-acetone solution of 10 was deep maroon, as expected, the solution of the pyran 9 was much less intensely coloured relative to that in commercial CDCl3 solution. The 1H NMR spectrum of 9 in d6-acetone displayed a singlet at δ 3.69 for the methoxy groups of the equivalent 4-methoxyphenyl units at C-2 and a singlet at δ 3.90 for the 6-methoxy group. The signal for 3-H appeared at δ 6.31 as a doublet (J = 9.6 Hz) coupled to 4-H (δ 6.77) and that for 5-H appeared as a singlet at δ 6.69. The 1H NMR spectrum of 10 in d6-acetone was quite different and displayed a singlet at δ 3.84, at δ 3.90 and at δ 4.02 assigned to the non-equivalent MeO groups. The protons of the allylidene side chain were unequivocally assigned by HSQC, HMBC, NOE and 1H–1H COSY experiments and appear at δ 7.38 (2′-H, partly obscured by other aromatic signals) and a doublet at δ 7.55 (1′-H); the magnitude of the coupling constant, J1′,2′ = 12.7 Hz, consistent with an s-trans conformation of the allylidene unit. The 13C NMR spectrum displayed the expected 2-C signal at δ 82.2 for the pyran 9 in d6-acetone and a low field signal at δ 183 confirmed the presence of the α,β-unsaturated carbonyl function in the 13C NMR spectrum of 10.
To contrast with the observed isomerisation behaviour of 9 and 10 in CDCl3 solution, treatment of a d6-acetone solution of pyran 9 with a catalytic amount of TFA resulted in a mixture of 9:10:11 in a ratio of ∼2:1:0.09, after standing over 2 h at rt (this composition remained essentially unchanged after 5 days) (ESI,† Fig. S20 and S21). Similarly treatment of a d6-acetone solution of dye 10 with a catalytic amount of TFA resulted in the formation of essentially the same equilibrium mixture which confirms the acid-mediated isomerisation but with the implication that the dye 10 is the thermodynamically favoured ring-opened form under the applied conditions.
The structure of 10 (Fig. 3) was unequivocally determined by X-ray diffraction from a single crystal grown by slow evaporation from EtOAc/hexane solution. Bond length alternation of the (E)-pentadienone unit was apparent with C3–C4 (1.486 (3) Å) and C12–C13 (1.436 (3) Å) proffering single bond like character with C3–C12 (1.362 (3) Å) and C13–C14 (1.355 (3) Å) possessing more double bond character; such data is comparable with that noted for 4-(3,3-diarylylallylidene)naphthalen-1(4H)-ones.9 In addition to the expected propeller-like arrangement of the geminal 4-methoxyphenyl rings, which arise as a consequence of the minimisation of ortho-H-atom interactions, there is a slight deviation of the allylidene unit from the plane of the naphthalenone (torsion angle 161.09° for the allylidene unit C3–C12–C13–C14); it is possible that this deviation is a consequence of the crystal packing in which the naphthalenone rings are arranged in an anti-planar array with a naphthalenone ring centroid spacing of 3.619 Å, as the consequence of a potential dipole–dipole interaction.13
 | ||
| Fig. 3 Crystallographic structure of 10 (thermal ellipsoids shown at 50% probability) and crystal packing. | ||
The photochromic response of 9 and the absorption spectrum of 10 were next examined in acetone solution. Dye 10 exhibits λmax at 496 nm and has a molar extinction coefficient (εm) of 2.64 × 104 mol−1 dm3 cm−1 the latter is comparable to εm of merocyanine dyes derived from 2-tetralone14 but lower than those derived from 1-naphthol.9 The absorption spectrum of 9 before UV irradiation confirms the weakly coloured solution with an absorbance of <0.05 a.u. at 507 nm, whereas upon UV irradiation for 30 s a deep maroon solution was observed with an absorption maximum at 507 nm (Fig. 4). Progressively longer irradiation times resulted in a small hypsochromic shift of λmax but with little if any increase in intensity after ca. 220 s of UV irradiation. The decay of the intensity of the absorption at 507 nm was measured as a function of time and the half-life (t1/2) was calculated to be 42.3 min at 20 °C (Table S1, ESI†).
 | ||
| Fig. 4 Absorption spectra (acetone) of naphthopyran 9 and dye 10. | ||
Of some significance was the fact that the absorption maximum of the coloured ring-opened form of pyran 9, resulting from 30 s of UV irradiation, was bathochromically shifted by 11 nm relative to that obtained for dye 10. It was thought that the different absorption maxima may be due to the photochemical ring-opening of the pyran 9 resulting in the alternate geometrical (Z)-isomer 11 of the isolated dye 10 [(E)-isomer] (Fig. 5).
 | ||
| Fig. 5 Relationship between pyran 9 and dyes 10 and 11. | ||
To obtain further insight on the differences between 10 and 11, we have used theoretical calculations (ESI† for details). For 10, the computed geometry agrees well with XRD with deviations of ca. 0.01–0.02 Å. Indeed, the experimental (theoretical) C3–C4, C3–C12, C12–C13 and C13–C14 bond lengths are 1.485 (1.480), 1.362 (1.377), 1.436 (1.421) and 1.355 (1.374) Å, respectively. In 11, these bond distances become 1.477, 1.383, 1.420 and 1.377 Å, respectively, which are similar but nevertheless show a slightly smaller bond length alternation, hinting at a marginally more delocalized structure. For both dyes, TD-DFT predicts a first dipole-allowed transition that can be mainly ascribed to a HOMO–LUMO contribution (see ESI† for representation). For 10, TD-DFT predicts a first excited-state at 541 nm, with an oscillator strength, f, of 0.86.15 For 11, theory predicts a small bathochromic (+11 nm or −0.05 eV, λmax = 552 nm) hypochromic (−0.05, f = 0.81) shift of the spectra, which seems to fit the experimental trends. To ascertain this result, we have also performed vibrationally resolved calculations of the optical spectra for each dye to reach theoretical estimates of εm and they also provided a weaker absorption for 11 than for 10.
To establish the presence of proposed isomer 11, a room temperature solution of 9 in d6-acetone was subjected to UV irradiation for ca. 30 s. Immediately upon cessation of irradiation the 1H NMR spectrum of the intensely coloured solution was acquired (Fig. 6). The 1H NMR spectrum revealed new signals at δ 3.84, 3.85 and 3.89 each accounting for a methoxy group and a singlet at δ 6.11 which is attributed to 3-H. The aromatic region of the spectrum was more complex and a signal of particular note was a low field doublet resonating at δ 8.65 with a coupling constant of 12.2 Hz which has been assigned to 2′-H; the partner signal for 1′-H resonates at δ 6.88 and is obscured by other aromatic signals. Longer irradiation (40 min) of a solution of 9 in d6-acetone resulted in the emergence of a further set of signals which were characteristic of the dye 10 (ESI,† Fig. S6 and S7). The 1H NMR spectrum of a sample of dye 10 after ca. 40 min of UV-irradiation was also examined. Interestingly, in this instance photo-isomerization of 10 resulted in a mixture of 9, 10 and 11 indicating that UV irradiation can affect the reverse geometrical isomerisation.
 | ||
| Fig. 6 1H NMR spectrum of naphthopyran 9 in d6-acetone after 30 s UV irradiation. | ||
We have also used theoretical calculations to analyse the spectra of 10 and 11 (see ESI†). By looking, at the three protons of the conjugated path shown in Fig. 5, it is apparent that the measured 10-to-11 isomeric effects on the chemical shifts are Δδ(3-H) −0.71 (δ6.11–6.82), Δδ(1′-H) −0.67 (δ6.88–7.55) and Δδ(2′-H) +1.27 (δ8.65–7.38) and theory reproduces these trends: Δδ(3-H) −0.62 (δ6.31–6.93), Δδ(1′-H) −0.99 (δ7.32–8.31) and Δδ(2′-H) +1.78 (δ9.45–7.67), respectively, therefore confirming the experimental assignment; the differences being within the expected error bars for DFT NMR simulations.
In order to assess the influence of an aryl ring on the properties of the pyran–merocyanine dye interconversion the reaction between 4-methoxy-1-naphthol and the propynol 8b was next undertaken. The crude reaction product resulting from this combination afforded two components after purification, the naphthopyran 12 (79%) and the propenone 13 (9%); no merocyanine dye analogous to 10 was observed (Scheme 2). The propenone 13, formed by a Meyer–Schuster rearrangement of 8b,16 was characterised by the presence of a signal at δ 192 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function in the 13C NMR spectrum and a singlet at δ 7.0 for the alkene proton in the 1H NMR spectrum. The naphthopyran 12 displayed no discernible photochromic response at ambient temperature however; a red-purple colour was discernible when a toluene solution chilled with solid CO2 was irradiated, but the colour faded immediately upon termination of irradiation. It is apparent that the presence of the 4-tolyl substituent favours the pyran 12, presumably as a consequence of increased steric interactions destabilising the merocyanine species.
O function in the 13C NMR spectrum and a singlet at δ 7.0 for the alkene proton in the 1H NMR spectrum. The naphthopyran 12 displayed no discernible photochromic response at ambient temperature however; a red-purple colour was discernible when a toluene solution chilled with solid CO2 was irradiated, but the colour faded immediately upon termination of irradiation. It is apparent that the presence of the 4-tolyl substituent favours the pyran 12, presumably as a consequence of increased steric interactions destabilising the merocyanine species.
 | ||
| Scheme 2 Naphthopyran 12 and propenone 13. | ||
In summary, this report constitutes the first example of the isolation and full characterisation of a merocyanine dye derived from a 2H-naphtho[1,2-b]pyran. Furthermore, examination of the absorption spectra of the merocyanine together with a sample of irradiated naphthopyran revealed that each isomeric merocyanine has a different absorption maximum as a consequence of the different geometry; a feature which is supported by TD-DFT calculations. The presence of low concentrations of acid favours the pyran ring-opening to afford the (E)-photomerocyanine. The foregoing features have implications for the study of the fading kinetics and applications of photochromic compounds in commercial ophthalmic systems.
We thank the EPSRC for access to the National Mass Spectrometry Service, Swansea. A. C.-E. and D. J. acknowledge the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of a Starting Grant (Marches-278845) and a recrutement sur poste stratégique, respectively. This research used resources of the GENCI-CINES/IDRIS, of the CCIPL (Centre de Calcul Intensif des Pays de Loire) and of a local Troy cluster.
Notes and references
- (a) U. Weigand and H. Zinner, WO Pat., 2014/009020, 2014 Search PubMed; (b) J. Takenada, J. Momoda, K. Teranishi, T. Takahashi, M. Sando and S. Izumi, US Pat., 2014/0054520, 2014 Search PubMed; (c) W. Xiao and B. Van Gemert, WO Pat., 2013/090220, 2013 Search PubMed; (d) A. Kumar, R. L. Yoest, C. Li, D. S. Jackson and H. Nguyen, US Pat., 2012/0120473, 2012 Search PubMed; (e) S. Aiken, J.-P. Cano, C. D. Gabbutt and B. M. Heron, WO Pat., 2008/028930, 2008 Search PubMed.
- (a) Y. Shimizu, S. Izumi and J. Momoda, WO Pat., 2013/042800, 2013 Search PubMed; (b) L. Sukhomlinova, T. Kosa, B. Taheri, T. White and T. Bunning, US Pat., 2013/0248350, 2013 Search PubMed; (c) D. A. Clarke, B. M. Heron, C. D. Gabbutt, J. D. Hepworth, S. M. Partington and S. N. Corns, US Pat., 2002/6387512, 2002 Search PubMed.
- (a) J. D. Hepworth and B. M. Heron, in Functional Dyes, ed. S.-H. Kim, Elsevier, Amsterdam, 2006, p. 85 Search PubMed; (b) B. Van Gemert, in Organic Photochromic and Thermochromic Compounds, Volume 1, Main Photochromic Families, ed. J. C. Crano and R. J. Guglielmetti, Plenum Press, New York, 1998, p. 111 Search PubMed.
- S. Delbaere and G. Vermeersch, J. Photochem. Photobiol., C, 2008, 9, 61 CrossRef CAS.
- S. Delbaere, B. Luccioni-Houze, C. Bochu, Y. Teral, M. Campredon and G. Vermeersch, J. Chem. Soc., Perkin Trans. 2, 1998, 1153 RSC.
- S. Delbaere, J.-C. Micheau and G. Vermeersch, Org. Lett., 2002, 4, 3143 CrossRef CAS PubMed.
- W. Zhao and E. M. Carreira, Org. Lett., 2003, 5, 4153 CrossRef CAS PubMed.
- R. Livingstone and M. C. Whiting, J. Chem. Soc., 1955, 3631 RSC.
- C. D. Gabbutt, B. M. Heron, A. C. Instone, D. A. Thomas, S. M. Partington, M. B. Hursthouse and T. Gelbrich, Eur. J. Org. Chem., 2003, 1220 CrossRef CAS.
- L. M. Carvalho, A. M. S. Silva, C. I. Martins, P. J. Coelho and A. M. F. Oliveira-Campos, Tetrahedron Lett., 2003, 44, 1903 CrossRef CAS.
- (a) J. E. Page, in Ann. Rep. on NMR Spectroscopy, ed. E. F. Mooney, Elsevier, Amsterdam, 1970, vol. 3, p. 149 Search PubMed; (b) S. Florio, G. Ingrosso and R. Sgarra, Tetrahedron, 1985, 41, 3091 CrossRef CAS; (c) J. A. Turner and W. Herz, J. Org. Chem., 1977, 42, 1657 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Elsevier, Oxford, 7th edn, 2013, p. 133 Search PubMed.
- Single crystal X-ray diffraction data was collected on a Bruker Venture diffractometer equipped with a graphite monochromated Cu(Kα) radiation source and a cold stream of N2 gas. Selected crystal data for 10: C28H24O4, M = 424.47, monoclinic, a = 17.9087 (7) Å, b = 13.6467 (6) Å, c = 17.9157 (7) Å, β = 94.4847 (19)°, V = 4365.1 (3) Å3, T = 150 K, space group C2/c, Z = 8, μ = 0.687 mm−1, 17375 measured reflections, 4119 independent reflections (Rint = 0.0555). The final R1 values were 0.0532 (I > 2σ(I)). The final wR(F2) values were 0.1554 (I > 2σ(I)). The final R1 values were 0.0657 (all data). The final wR(F2) values were 0.1668 (all data). The goodness of fit on F2 was 1.062. Largest peak and hole 0.267 and −0.291 e Å−3. See CCDC 1000252.
- C. D. Gabbutt, J. D. Hepworth, B. M. Heron, S. M. Partington and D. A. Thomas, Dyes Pigm., 2001, 49, 65 CrossRef CAS.
- This reasonably fits the experimental value of 496 nm and corresponds to an error of 0.21 eV, typical of TD-DFT. See a recent review on TD-DFT benchmarks: A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019 CrossRef CAS.
- (a) C. D. Gabbutt, B. M. Heron, C. Kilner and S. B. Kolla, Org. Biomol. Chem., 2010, 8, 4874 RSC; (b) D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures, characterization data and copies of NMR and mass spectra for all new compounds, computed Cartesian coordinates, frontier molecular orbitals and vibronic spectra. CCDC 1000252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03435j |
| This journal is © The Royal Society of Chemistry 2014 |