
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electron transfer series of octahedral chromium complexes containing a redox non-innocent α-diimine ligand†
Jingmei
Shen‡
a,
Glenn P. A.
Yap
a,
William E.
Barker IV
b,
William E.
Geiger
b and
Klaus H.
Theopold
*a
aDepartment of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: theopold@udel.edu
bDepartment of Chemistry, University of Vermont, Burlington, VT 05405, USA
First published on 24th July 2014
Abstract
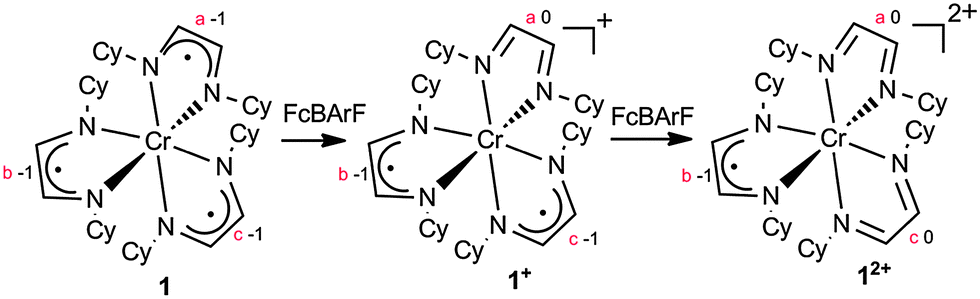
An electron-transfer series of octahedral α-diimine complexes [(HLCy)3Cr]n+(BARF)n (n = 2, 1, 0) has been synthesized and crystallographically characterized. Cyclic voltammetry indicated additional formation of [(HLCy)3Cr]3+. The molecular structures suggested that all redox processes were ligand-based. Magnetic moments were consistent with spin ground states of S = 0 for [HLCy3Cr]0, S = 1/2 for [HLCy3Cr]+1, and S = 1 for [HLCy3Cr]+2. The experimental data is consistent with chromium maintaining the +III oxidation state throughout, while being coordinated by varying numbers of neutral diimines (HLCy) and diimine radical anions (HLCy˙−).
As part of our wide-ranging exploration of open-shell chromium organometallics,1 we have recently employed α-diimines (aka 1,4-diazadienes) as ancillary ligands.2 We were particularly attracted by the redox activity of these ligands, which might be expected to ameliorate the strictures imposed by oxidation state changes during catalytic cycles (e.g. CrI/CrIII in the selective oligomerization of ethylene).3 Three oxidation states of the α-diimine ligands have been identified, namely the neutral diimine (L), the radical monoanions (L˙−), and the two-electron-reduced enediamide (L2−).4 The assignment of ligand oxidation states is informed by their detailed structural parameters, which change in a predictable way; however, spectroscopic and computational techniques are also often employed.
The electronic flexibility of α-diimine ligands enables the preparation of molecules with unusual atomic and electronic structures. A representative example from our laboratory was the serendipitous discovery of dinuclear [HLiPrCr]2 (HLiPr = N,N′-bis(2,6-diisopropylphenyl-1,4-diazadiene)),5 which features a rather short Cr–Cr quintuple bond between Cr(I) (d5) ions coordinated by two singly reduced bridging α-diimines (HLiPr˙−) and exhibits notable reactivity with a wide variety of small molecules.6 In an extension of this chemistry, we encountered the deceptively simple octahedral complex (HLCy)3Cr (1, HLCy = N,N′-bis(cyclohexyl)-1,4-diazadiene). In years past, 1 might have been described as a low-spin Cr(0) (d6) complex containing neutral diimine ligands. However, in the age of lost ligand innocence,7 all indications are that 1 is best understood as a Cr(III) (d3, S = 3/2) complex, with strong antiferromagnetic coupling to three HLCy˙− radical anions (S = 1/2).8 This description is consistent with that of analogous compounds, i.e. those containing ligands such as 2,2′-bipyridine (bpy), 2,2′,6′,2′′-terpyridine (terpy),9 dithiolene,10 α-catecholates,11 and others. Herein we report on the redox chemistry of 1, which is extensive and appears entirely ligand based.
The synthesis and characterization of 1 has been described;8 it is an air sensitive material that can easily be oxidized (see Scheme 1). Thus treatment of a brown Et2O solution of 1 with 1.0 equivalent of FcBARF (Fc+ = ferricenium, BARF− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) yielded a deep purple solution, from which [(HLCy)3Cr]BARF ([1+]BARF) was isolated in 92% yield after recrystallization from Et2O/toluene. In contrast to 1, solid [1+]BARF appears to be stable in air; its structure has been determined by X-ray diffraction and the result is shown in Fig. 1, with selected bond distances listed in Table 1.
 | ||
| Scheme 1 The electron-transfer series for complexes 1–12+. | ||
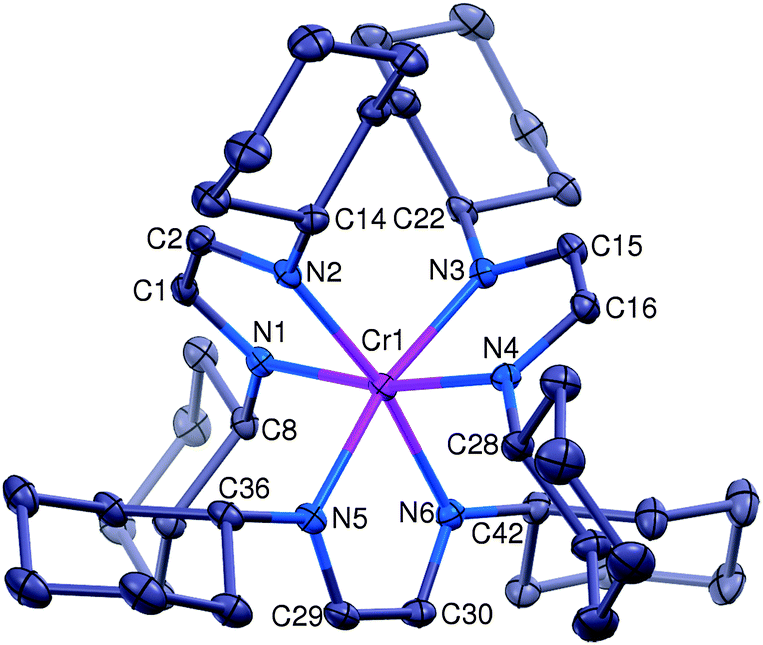 | ||
| Fig. 1 The structural diagram of the [(HLCy)3Cr]+ cation (1+) at 30% probability. The BARF− counterion, one toluene solvent molecule and all hydrogen atoms have been omitted for clarity. | ||
| Electrolyte/medium | E 1/2 (13+/12+) (V) | E 1/2 (12+/1+) (V) | E 1/2 (1+/1) (V) |
|---|---|---|---|
| CH2Cl2/0.05 M [NBu4][B(C6F5)4] | 0.78 | −0.27 | −1.56 |
| THF/0.1 M [NBu4][PF6] | — | −0.50 | −1.37 |
Further oxidation of 1+ proved feasible. Synthetic access to the green [(HLCy)3Cr][BARF]2 ([12+](BARF)2) can be had either by the oxidation of 1 with 2.0 equivalents of FcBARF or by the reaction of [1+]BARF with 1.0 equivalent of FcBARF in Et2O. Recrystallization from methylene chloride at −30 °C afforded air-stable green plates of [12+](BARF)2 in 70% yield. The solid-state structure of 12+ has been determined; it is similar in its gross features to those of 1 and 1+ (see Fig. S1, ESI†). The isolation of a characterizable [(HLCy)3Cr]3+ salt was not met with success, however. Strong oxidants, such as NO+ and [N(C6H4Br-4)3]+, were explored in attempts to oxidize 12+. These reactions afforded yellow precipitates, which were very sparingly soluble in common solvents such as THF, acetone, methanol or water. While these solids may indeed contain the desired trication (see below), their limited solubility has so far prevented further characterization.
The electrochemistry of 1n+ was investigated by cyclic voltammetry (CV) and bulk electrolysis carried out on solutions of [1+]BARF. In both THF and dichloromethane, 1+ undergoes a single one-electron reduction (to 1) and a pair of one-electron oxidations (to 12+ and 13+). Fig. 2 is representative of the observed voltammetry, which is consistent with the presence of a four-membered electron transfer series having the E1/2 potentials given in Table 1 (the oxidation of 12+ was not observed in THF).
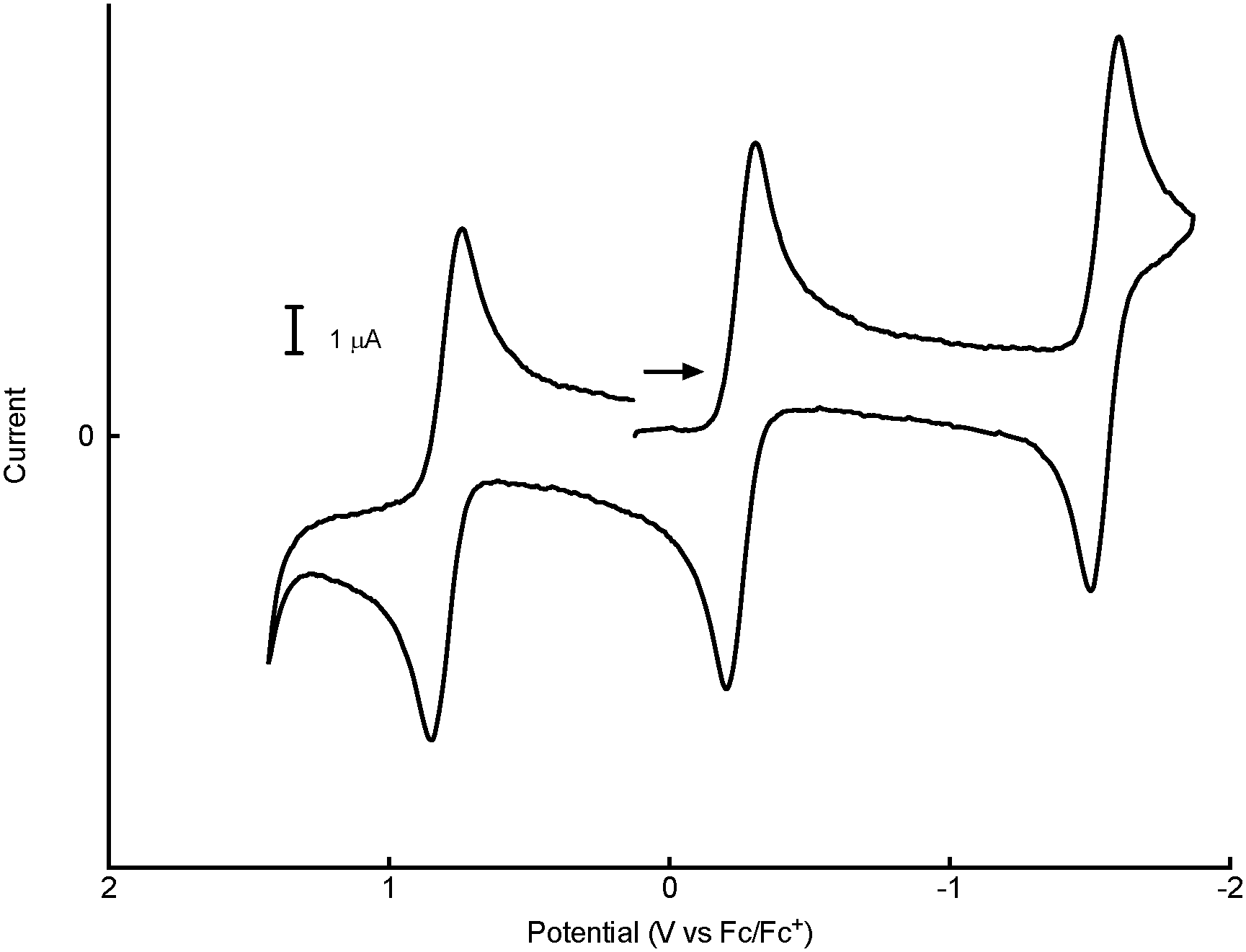 | ||
| Fig. 2 Cyclic voltammogram of 1 mM 12+ in CH2Cl2/0.05 M [NBu4][B(C6F5)4] at a 2 mm glassy carbon electrode, scan rate 0.2 V s−1. This was formed by anodic bulk electrolysis of a 1 mM solution of 1+[BARF] at Eapplied = 0.3 V. The CV scan was initiated negatively from 0 V, as indicated by the arrow. | ||
Bulk electrolysis experiments carried out in both solvents confirmed the one-electron nature of the reactions and the thermal stabilities of the various redox states. For example, anodic electrolysis of 1+ in dichloromethane/[NBu4][B(C6F5)4] at Eappl = 0.1 V resulted in clean conversion of a purple solution of the monocation to the blue-green solution of 12+. Moving Eappl to 1.1 V converted 12+ to a yellow-orange solution of 13+. Reversing this sequence regenerated 1+, apparently without loss of the compound. One-electron reduction of 1+ was carried out in THF/[NBu4][PF6] at Eappl = −1.7 V, giving the neutral complex 1, which was subsequently re-oxidized to 1+ at Eappl = −1 V. The three electron-transfer reactions involving 13+/12+/1+/1 are highly chemically reversible under our experimental conditions.
The observation of three reversible redox processes raises the question of the electronic structure and formal oxidation state of chromium of the various members of this electron transfer series. The most reduced compound, 1, has unambiguously been assigned an electronic structure represented by (L˙−)3CrIII;8 it seems highly improbable that its oxidation should result in a lowering of the metal's oxidation state below +III. Accordingly, there remains the question whether the successive oxidations remove electrons from the metal or from the ligands. To answer this question we turn to a consideration of the structural details of these complexes. We note that all three molecules feature octahedral coordination of chromium. Given the pronounced stabilization of the d3 electronic configuration in an octahedral ligand field, and the known tendency for chromium in higher oxidation states to adopt lower coordination numbers, this is already a hint. Selected interatomic distances for 1, 1+, and 12+ are listed in Table 2.
| Compound | 1 | 1 + | 12 + |
|---|---|---|---|
| Subscripts a, b and c refer to symmetry unique diimine ligands. | |||
| C–Ca | 1.390(3) | 1.408(4) | 1.444(8) |
| C–Na | 1.333(2), 1.332(2) | 1.314(4), 1.316(4) | 1.291(6), 1.300(7) |
| Cr–Na | 2.0377(16), 2.0418(14) | 2.025(3), 2.024(2) | 2.088(4), 2.105(4) |
| C–Cb | 1.397(5) | 1.414(8) | |
| C–Nb | 1.318(4), 1.318(4) | 1.324(7), 1,299(6) | |
| Cr–Nb | 2.019(3), 2.023(3) | 2.061(4), 2.048(4) | |
| C–Cc | 1.397(4) | 1.431(8) | |
| C–Nc | 1.318(4), 1.316(4) | 1.309(7), 1.287(7) | |
| Cr–Nc | 2.026(3), 2.029(3) | 2.046(4), 2.079(4) | |
| C–Cavg. | 1.390(3) | 1.401(5) | 1.430(8) |
| C–Navg. | 1.332(2) | 1.317(4) | 1.302(7) |
| Cr–Navg. | 2.0398(16) | 2.024(3) | 2.071(4) |
We note that the Cr–N distances in all three compounds vary within a very narrow range (2.019(3)–2.105(4) Å), without any discernable trend. As higher oxidation states tend to engender shorter metal–ligand distances, this too suggests that the oxidation state of the metal is not changing. For this to be true, successive oxidations must remove electrons from the coordinated diimine radical anions, which should result in systematic changes of the C–C and C–N bond distances of the ligand backbones. Although both 1+ and 12+ lack threefold crystallographic symmetry, rendering the ligands inequivalent, their metric data do not support the unambiguous identification of one (or two) neutral diimines in 1+ (or 12+), respectively. In other words, the C–C and C–N distances of the individual ligands in any given compound do not differ significantly. In principle, this may be due to positional disorder in the crystal, or it might indicate effective delocalization of the available electrons over all three ligands. The present structural data cannot distinguish between these two alternatives. Interestingly, DFT calculations on models for 1+ and 12+ are consistent with delocalization in the former (Fig. S12, ESI†) and localization in the latter (Fig. S13, ESI†). In any event, inspection of the average distances (see Table 2, bottom) reveals a gradual lengthening of the C–C bonds and a concomitant foreshortening of the C–N bonds in the order 1 → 1+ → 12+. Thus, in this order the ligands look on average less like radical anions (L˙−) and more like neutral diimines (L). To summarize, the structural data are consistent with formulations of 1+ as [(L˙−)2(L)CrIII]+ and 12+ as [(L˙−)(L)2CrIII]2+.
The electronic spectra are shown in Fig. 3. The intense high energy bands are likely π → π* transition of the ligands, while the weak transition in the visible region may be attributed to d → d transitions of the Cr(III) ion. Most conspicuous are the broad bands in the near IR (1389 nm for 1+, 923 and 1006 nm for 12+), which are absent from the spectrum of 1; these are tentatively assigned as ligand-to-ligand intervalence charge-transfer bands associated with the presence of ligands in different redox states on the same metal center.9d
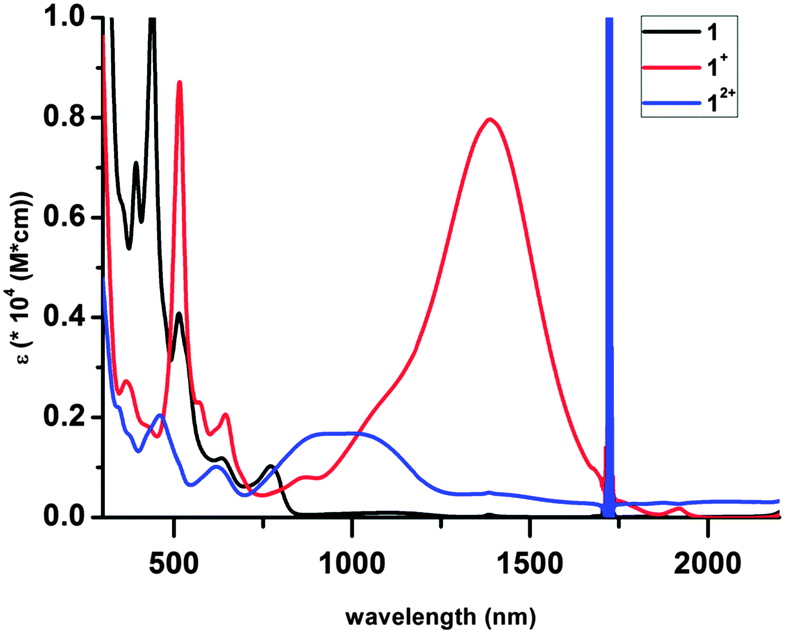 | ||
| Fig. 3 Electronic spectra of complexes 1–12+ in THF. | ||
To further characterize the electronic structures of this series of compounds, we have carried out magnetic measurements. The temperature dependencies of the effective magnetic moments of all three complexes are depicted in Fig. 4. Both [1+](BARF) [1](BF4)2 are simple paramagnets, whose molar magnetic susceptibilities can be fit with Curie–Weiss expressions. Accordingly, their effective magnetic moments are temperature independent over the whole temperature range of 50–350 K. Their respective values are consistent with spin ground states of S = 1/2 for 1+ and S = 1 for 12+. These presumably result from strong antiferromagnetic coupling between the central Cr(III) ion (S = 3/2) and two or one diimine radical anions (S = 1/2), respectively. Extending this rationale to 1 would predict diamagnetism (ground state S = 0) for this complex. However, as can be seen in Fig. 4, 1 actually exhibits a rather small, and temperature dependent magnetic moment. Indeed, this residual paramagnetism was also evident in the 1H NMR spectrum of 1. While its resonances were found in the ‘normal’ shift range (δ 1–10 ppm), the room temperature spectrum (in THF-d8) exhibited a broad resonance at ca. 4.7 ppm, which split into two sharp resonances upon cooling. This apparent ‘coalescence phenomenon’ was ultimately traced to significant temperature dependencies of the chemical shifts of the protons on the backbone of the diimine and the cyclohexyl protons adjacent to the nitrogen, which are accidentally isochronous at room temperature (see ESI† for more detail). The residual paramagnetism of 1 can be interpreted as a result of antiferromagnetic coupling between the metal and the ligands that is not strong enough to entirely preclude the occupation of the first excited spin state (S = 1).9d
 | ||
| Fig. 4 Temperature dependence of the magnetic moments of 1–12+. | ||
We have shown that octahedral tris(diimine) chromium complexes form an extensive electron-transfer series of the composition [(HLCy)3Cr]n+ (n = 3, 2, 1, 0). At least three, and most likely all four of these complexes feature chromium in its highly favored +III oxidation state. The redox processes are wholly ligand based, thus confirming that α-diimines belong to a growing class of non-innocent ligands that extend the redox capacity of metal complexes. This should prove useful in catalytic cyles involving multi-electron redox processes.
This work was supported by the NSF (CHE-0911081 to KHT).
Notes and references
- (a) K. H. Theopold, Acc. Chem. Res., 1990, 23, 263 CrossRef CAS; (b) R. A. Heintz, S. Leelasubcharoen, L. M. Liable-Sands, A. L. Rheingold and K. H. Theopold, Organometallics, 1998, 17, 5477 CrossRef CAS; (c) L. MacAdams, G. Buffone, C. Incarvito, A. Rheingold and K. Theopold, J. Am. Chem. Soc., 2005, 127, 1082 CrossRef CAS PubMed; (d) W. H. Monillas, G. P. A. Yap and K. H. Theopold, Angew. Chem., Int. Ed., 2007, 46, 6692 CrossRef CAS PubMed; (e) W. H. Monillas, G. P. A. Yap, L. A. MacAdams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090 CrossRef CAS PubMed.
- (a) K. A. Kreisel, G. P. A. Yap and K. H. Theopold, Eur. J. Inorg. Chem., 2012, 520 CrossRef CAS; (b) K. A. Kreisel, G. P. A. Yap and K. H. Theopold, Inorg. Chem., 2008, 47, 5293 CrossRef CAS PubMed.
- (a) J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641 CrossRef CAS PubMed; (b) D. F. Wass, Dalton Trans., 2007, 816 RSC; (c) T. Agapie, Coord. Chem. Rev., 2011, 255, 861 CrossRef CAS PubMed; (d) W. H. Monillas, J. F. Young, G. P. A. Yap and K. H. Theopold, Dalton Trans., 2013, 42, 9198 RSC.
- (a) N. Muresan, C. C. Lu, M. Ghosh, J. C. Peters, M. Abe, L. M. Henling, T. Weyhermüller, E. Bill and K. Wieghardt, Inorg. Chem., 2008, 47, 4579 CrossRef CAS PubMed; (b) M. Ghosh, S. Sproules, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2008, 47, 5963 CrossRef CAS PubMed; (c) N. Muresan, K. Chlopek, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2007, 46, 5327 CrossRef CAS PubMed; (d) C. C. Lu, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 2008, 130, 3181 CrossRef CAS PubMed; (e) C. C. Lu, S. DeBeer George, T. Weyhermüller, E. Bill, E. Bothe and K. Wieghardt, Angew. Chem., Int. Ed., 2008, 47, 6384 CrossRef CAS PubMed.
- K. A. Kreisel, G. P. A. Yap, O. Dmitrenko, C. R. Landis and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 14162 CrossRef CAS PubMed.
- (a) J. M. Shen, G. P. A. Yap, J. P. Werner and K. H. Theopold, Chem. Commun., 2011, 47, 12191 RSC; (b) J. M. Shen, G. P. A. Yap and K. H. Theopold, J. Am. Chem. Soc., 2014, 136, 3382 CrossRef CAS PubMed; (c) J. Shen, G. P. A. Yap and K. H. Theopold, Chem. Commun., 2014, 50, 2579 RSC.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed.
- M. Wang, J. England, T. Weyhermüller, S. L. Kokatam, C. J. Pollock, S. DeBeer, J. M. Shen, G. P. A. Yap, K. H. Theopold and K. Wieghardt, Inorg. Chem., 2013, 52, 4472 CrossRef CAS PubMed.
- (a) C. C. Scarborough, S. Sproules, C. J. Doonan, K. S. Hagen, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2012, 51, 6969 CrossRef CAS PubMed; (b) C. C. Scarborough, K. M. Lancaster, S. DeBeer, T. Weyhermüller, S. Sproules and K. Wieghardt, Inorg. Chem., 2012, 51, 3718 CrossRef CAS PubMed; (c) J. England, C. C. Scarborough, T. Weyhermüller, S. Sproules and K. Wieghardt, Eur. J. Inorg. Chem., 2012, 4605 CrossRef CAS; (d) C. C. Scarborough, S. Sproules, T. Weyhermüller, S. DeBeer and K. Wieghardt, Inorg. Chem., 2011, 50, 12446 CrossRef CAS PubMed; (e) C. C. Scarborough and K. Wieghardt, Inorg. Chem., 2011, 50, 9773 CrossRef CAS PubMed.
- (a) P. Banerjee, S. Sproules, T. Weyhermüller, S. D. George and K. Wieghardt, Inorg. Chem., 2009, 48, 5829 CrossRef CAS PubMed; (b) S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2011, 255, 837 CrossRef CAS PubMed.
- (a) C. Benelli, A. Dei, D. Gatteschi, H. U. Güdel and L. Pardi, Inorg. Chem., 1989, 28, 3089 CrossRef CAS; (b) R. R. Kapre, E. Bothe, T. Weyhermüller, S. D. George, N. Muresan and K. Wieghard, Inorg. Chem., 2007, 46, 7827 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparative and crystallographic and spectroscopic data. CCDC 1000091 and 1000092. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03332a |
| ‡ Current address: Optimal CAE Inc., Plymouth, MI 48170, USA. |
| This journal is © The Royal Society of Chemistry 2014 |