
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chemical switch for controlling viral infectivity†
Maximilian
Hörner‡
abc,
Beate
Kaufmann‡
a,
Gabriella
Cotugno
d,
Ellen
Wiedtke
d,
Hildegard
Büning‡
e,
Dirk
Grimm‡
d and
Wilfried
Weber
*abc
aFaculty of Biology, University of Freiburg, Schänzlestrasse 1, 79104 Freiburg, Germany. E-mail: wilfried.weber@biologie.uni-freiburg.de; Fax: +49 761 203 97660; Tel: +49 761 203 97654
bBIOSS – Centre for Biological Signalling Studies, University of Freiburg, Schänzlestrasse 18, 79104 Freiburg, Germany
cSGBM – Spemann Graduate School of Biology and Medicine, University of Freiburg, Albertstrasse 19A, 79104 Freiburg, Germany
dHeidelberg University, Cluster of Excellence CellNetworks, Department of Infectious Diseases, Virology, Im Neuenheimer Feld 267, 69120 Heidelberg, Germany
eCenter for Molecular Medicine Cologne (CMMC), University of Cologne, CMMC Research Building, Robert-Koch-Str. 21, 50931 Cologne, Germany
First published on 17th July 2014
Abstract
Chemically triggered molecular switches for controlling the fate and function of biological systems are fundamental to the emergence of synthetic biology and the development of biomedical applications. We here present the first chemically triggered switch for controlling the infectivity of adeno-associated viral (AAV) vectors.
Chemically triggered molecular switches to control biological processes represent a cornerstone technology in the recent emergence of synthetic biology.1 Such chemical switches allow nowadays to control at will biological processes on every level of the cellular signaling cascade such as receptor activation,2 modulation of signaling pathways,3 gene expression4 or protein degradation.5 The time-resolved chemical control of cellular function has provided unmatched insight into pathological and physiological processes6 and has opened novel opportunities in drug discovery, manufacturing of biopharmaceuticals, gene therapy and tissue engineering.7
So far, such chemically controlled synthetic biological switches have been reported which require the prior introduction of transgenes into the cell either by non-viral or viral vector means.8 However, no system has been described to date, in which a conditionally controlled transgene transfer itself was used as a control point for the expression of a (therapeutic) target gene. Controlling transgene transfer to cells in a switchable manner could be used to avoid off-target expression and thus lower the risk of side effects. Due to their high efficiency and amenability to genetic engineering, viral vectors are preferred vehicles for transferring foreign DNA into mammalian cells.9 In particular, vectors based on the adeno-associated virus (AAV) have become popular and have recently been approved as the first gene therapeutics in the EU.10 In this study we describe a novel strategy to control infectivity. Specifically, we report – using AAV vectors as an example – the first small molecule-triggered chemical switch that defines viral infectivity.
The chemically switchable AAVSWITCH is based on an engineered serotype 2 viral capsid and on an adapter protein mediating inducible infectivity. The engineered capsid contains two modifications: (i) the capsid's natural infectivity was eliminated by mutating two key residues for AAV's primary receptor binding on target cells, a heparin sulfate proteoglycan (HSPG) structure (mutations R585A and R588A11 in the viral capsid proteins VP1, VP2 and VP3) and (ii) the human FK-binding protein (FKBP)12 was fused at the amino-terminal end to the viral capsid protein VP2 since it was previously shown that large insertions are tolerated at this position.13 The adapter protein consists of a modified FKBP–rapamycin binding (FRB) domain of mTOR14 fused to the fluorescent protein mCherry (for visualization) and a designed ankyrin repeat protein (DARPin) specific for the human epidermal growth factor receptor (DARPinEGFR17). In this configuration, addition of the non-immunosuppressive and commercially available rapamycin structural analog AP2196718 induces heterodimerization of the FRB–FKBP domains and thus the recruitment of cell-binding DARPinEGFR to the viral capsid surface, thereby restoring binding of the capsid to the cell surface and subsequent infection. However, in the absence of AP21967 the capsid cannot bind to the cell surface, thus preventing infection (Scheme 1).
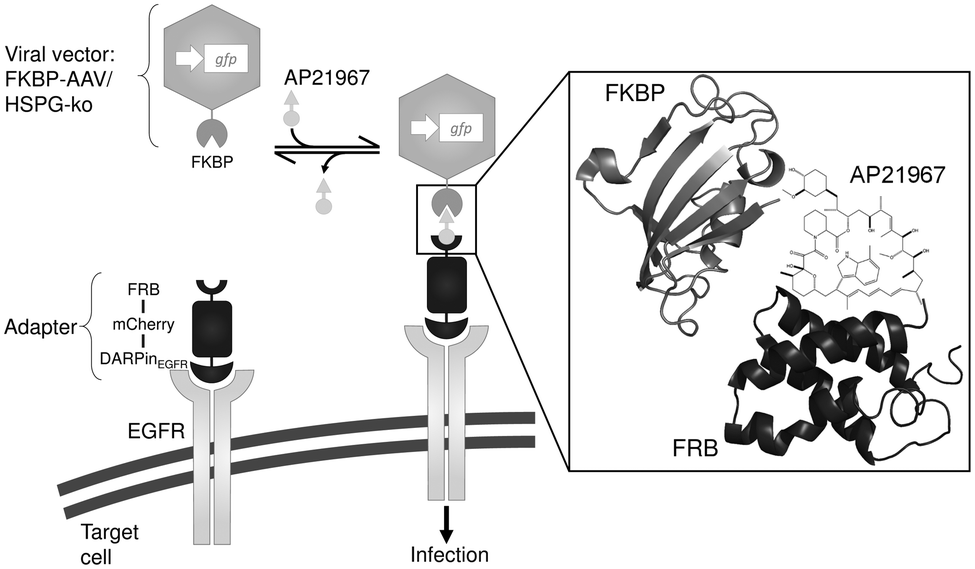 | ||
| Scheme 1 Schematic representation of the chemically switchable adeno-associated viral vector. Adeno-associated virus serotype 2 (AAV-2) vector particles were equipped with a chemical switch consisting (i) of the FK-binding protein (FKBP) fused to the viral capsid protein VP2 (FKBP–AAV) and (ii) of an FKBP–rapamycin binding (FRB) domain fused to the fluorescent protein mCherry and a DARPin specifically targeting the epidermal growth factor receptor (EGFR) on the surface of mammalian cells (DARPinEGFR). Upon addition of the rapamycin analog AP21967, binding of FKBP to FRB is induced leading to recruitment of the AAV-2 particle to the cell and subsequent infection. In order to prevent unspecific infection, the natural tropism of AAV-2 towards its primary receptor heparan sulfate proteoglycan (HSPG) was ablated by amino acid substitutions (R585A and R588A) in the viral capsid proteins VP1, VP2 and VP3 (HSPG-ko). Schematic representations of FRB and FKBP were generated using PyMOL with PDB files 2GAQ15 and 2PPN,16 respectively. | ||
For the synthesis of the adapter protein, a bacterial expression vector was constructed encoding (from N- to C-terminus) the FRB domain (FRB consists of amino acids 2021–2113 of the human FRAP in which the threonine at amino acid 2098 was mutated to leucine, to accommodate the chemical substitution that prevents AP21967 binding to wild-type FRAP18b) fused via a glycine–serine linker to the red fluorescent protein mCherry and to DARPinEGFR. A hexahistidine tag was fused to the carboxy terminus for purification. The protein was produced in E. coli (titer: ∼100 mg protein per 1 L culture) and purified by immobilized metal ion affinity chromatography (IMAC, see Fig. S1, ESI†). The functionality of the DARPinEGFR domain was confirmed by incubating the adapter protein with the EGFR-overexpressing human epidermoid carcinoma A-431 cell line for 1 h and the subsequent visualization of cell-bound mCherry via fluorescence microscopy and flow cytometry (Fig. S2, ESI†). Confocal microscopy analysis revealed strong red fluorescence at the membrane and partly in the cytoplasm likely caused by EGFR internalization.19 However, cells treated with an adapter protein variant lacking the DARPinEGFR domain did not show red fluorescence thereby confirming specific DARPinEGFR-mediated cell binding (Fig. S2a, ESI†). Quantitative analysis of adapter-cell interaction by flow cytometry confirmed the microscopy observations and revealed its dose-dependency (Fig. S2b, ESI†).
In order to confirm the functionality of the FRB domain in the adapter protein, we evaluated its AP21967-dependent binding to an FKBP protein fused to the yellow fluorescent protein mVenus (for synthesis of the FKBP-mVenus protein, see the ESI† and Fig. S1). To this aim, equimolar amounts of the adapter and FKBP–mVenus were mixed, incubated with or without 5-fold molar excess of AP21967 for 1 h and subsequently analyzed by size exclusion chromatography (Fig. S3, ESI†). In the absence of AP21967, FRB–mCherry–DARPinEGFR and FKBP–mVenus were both separated as monomers, while in the presence of AP21967 both proteins eluted as heterodimers thereby confirming functionality of FRB (Fig. S3, ESI†).
Viral vector particles with an engineered capsid were produced by the adenovirus helper-free AAV serotype-2 packaging system (Fig. 1a).20 In order to eliminate natural infectivity, arginines 585 and 588 of the viral capsid proteins VP1, VP2 and VP3 were mutated to alanine (HSPG-ko mutation).11 Furthermore, a silent mutation was introduced within the VP2 start codon T138 of the rep-cap plasmid to prevent translation of the unmodified VP2 protein (plasmid pRCVP2koA). The gene encoding the fusion protein FKBP–VP2/HSPG-ko was expressed in trans from a separate vector (plasmid pMH156).13,21 In order to visualize viral infection, a vector genome was used encoding an expression cassette for the green fluorescent protein (plasmid pCMVgfp). These plasmids were transfected into human embryonic kidney cells (HEK-293T) together with an adenovirus-derived helper plasmid (pHelper), and viral vector particles were isolated by cell lysis and subsequent purification by ultracentrifugation on an iodixanol density gradient. The viral titer was determined by quantitative genomic PCR and revealed 4.1 × 1011 genomic copies per mL vector stock.
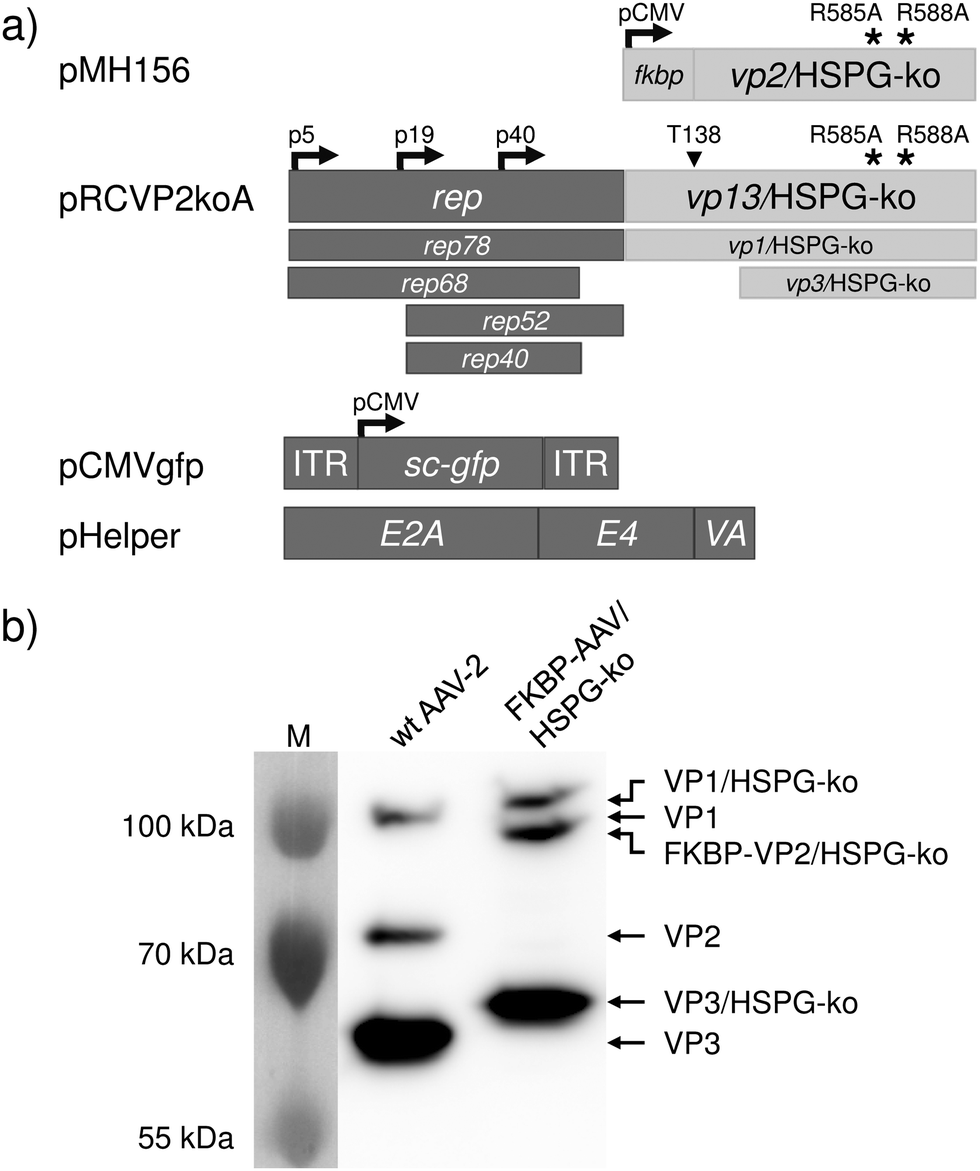 | ||
| Fig. 1 Synthesis and characterization of the FKBP–AAV/HSPG-ko particles. (a) Plasmids used for the synthesis of FKBP–AAV/HSPG-ko particles. Plasmid pMH156 encodes FKBP fused to the viral capsid protein VP2 harboring the R585A and R588A mutations for ablating the natural tropism for HSPG. Plasmid pRCVP2koA2 encodes the four non-structural proteins required for replication (Rep78, Rep68, Rep52 and Rep40) as well as the structural proteins VP1, VP3 and AAP (assembly activating protein, not shown). The overlapping open reading frames of the capsid proteins further contain the mutations R585A and R588A for ablating the natural tropism as well as a silent mutation within the VP2 start codon T138 in order to prevent expression of wildtype VP2. Plasmid pCMVgfp harbors a self-complementary AAV genome22 encoding green fluorescent protein (sc-GFP) under the control of the CMV promoter. Plasmid pHelper provides adenoviral genes E2A,23 E424 and VA25 required for the production of functional AAV vector particles. (b) Production and characterization of FKBP–AAV/HSPG-ko particles. FKBP–AAV/HSPG-ko were produced by co-transfecting the plasmids shown in (a) into HEK-293T cells. Following purification by density gradient centrifugation, viral vector particles were analyzed by 8% SDS-PAGE and visualized by Western blot analysis. As control, AAV-2 vectors with wildtype (wt) capsid were used. Calculated molecular sizes: VP1 and VP1/HSPG-ko: 82 kDa; VP2: 67 kDa; FKBP–VP2/HSPG-ko: 79 kDa; VP3 and VP3/HSPG-ko: 60 kDa. Abnormal mobility of AAV capsid proteins in SDS-PAGE is in accordance with previous observations26 and the reduced mobility of the R585A and R588A containing capsid proteins was already shown.21 ITR, inverted terminal repeat; M, molecular weight marker; p5, p19, p40, adeno-associated viral promoters; pCMV, human cytomegalovirus promoter. | ||
Incorporation of the engineered proteins into the viral capsid was analyzed by SDS-PAGE and Western blotting revealed the expected presence of the VP1 and VP3 HSPG-ko variants and of the FKBP–VP2/HSPG-ko fusion protein (Fig. 1b).
Based on the successful synthesis and characterization of the adapter protein and the engineered viral capsid, we evaluated the functionality of AAVSWITCH. To this aim, 4.8 × 104 A-431 cells were seeded per well (volume: 600 μl), incubated overnight and subsequently supplemented with 10−7 M adapter protein, 3.3 × 104 viral vector particles per cell (based on genomic copy number) and optionally with 1 μM AP21967. After 48 h, infection was analyzed by visualizing the expression of the vector-encoded reporter gene gfp by confocal microscopy (Fig. 2a). Under both conditions (with and without AP21967) binding of the adapter protein to cells was confirmed by membrane- and cytoplasm-localized red fluorescence. However, only in the presence of AP21967, green fluorescent cells were observed indicating the functionality of the chemically switchable viral vector AAVSWITCH (Fig. 2a).
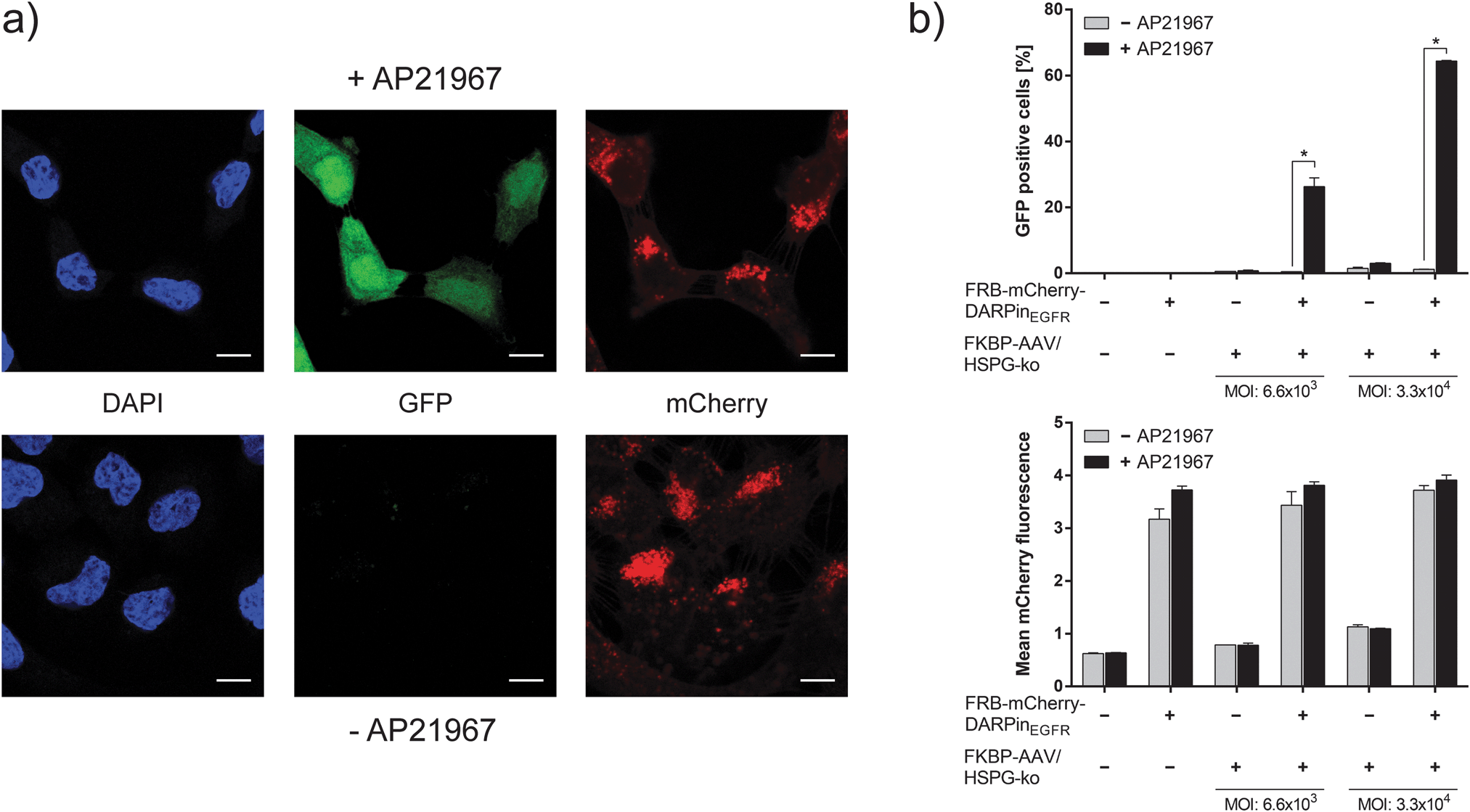 | ||
Fig. 2 Chemically switchable viral infection of mammalian cells. (a) Chemically switchable infection. 9.6 × 104 A-431 cells cultivated in 600 μl medium were supplemented with FRB–mCherry–DARPinEGFR (final concentration 10−7 M) and 3.2 × 109 FKBP–AAV/HSPG-ko particles (based on genomic copies (gc), MOI (genomic particles per cell): 3.3 × 104). Cells were cultivated in the absence or presence of 1 μM AP21967 for 48 h prior to confocal imaging of GFP and mCherry. Cell nuclei were visualized by DAPI staining. Scalebar, 10 μm. (b) Quantitative characterization of the chemically switchable infection. 1.6 × 104 A-431 cells cultivated in 100 μl medium were supplemented with FRB–mCherry–DARPinEGFR (final concentration 10−7 M) and 1.1 × 108 or 5.3 × 108 FKBP–AAV/HSPG-ko particles (MOI: 6.6 × 103 or 3.3 × 104). The samples were cultivated for 48 h in the absence or the presence of 1 μM AP21967 prior to flow cytometry analysis for quantifying GFP and mCherry fluorescence. A minimum of 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were analyzed per condition. Data are means ± standard deviation (n = 3). *P < 0.005 (two-sided t-test). 000 cells were analyzed per condition. Data are means ± standard deviation (n = 3). *P < 0.005 (two-sided t-test). | ||
The performance of the chemically switchable infection was quantified by flow cytometry in the absence and presence of the adapter protein and using different vector doses (Fig. 2b and Fig. S4, ESI†). In the absence of the adapter, only background GFP levels were detected reflecting the efficacy of the HSPG-ko mutation to ablate natural viral tropism. In the presence of the adapter protein and the viral vector particles but without AP21967, GFP levels similar to those of the adapter-lacking controls were observed. However, upon addition of the chemical inducer AP21967 (1 μM), a more than 50-fold increase in GFP levels was observed thereby confirming the previous qualitative microscopy analysis (Fig. 2a) and the functionality of the AAVSWITCH system. The relative gene-delivery efficiency of AAVSWITCH in the AP21967-induced state was in the same range (75–85%, Fig. S5, ESI†) as that of AAV-2 vectors with unmodified capsids using the same number of viral vector particles per cell.
To show that AAVSWITCH is specific for EGFR-overexpressing cells, the system was tested with five other cell lines reported to express EGFR at different levels (Fig. S5 and S6, ESI†). It could be observed that the AAVSWITCH system infects cells with high27 (A-431) or medium28 (A549, HeLa, MDA-MB-231) EGFR expression in an AP21967-dependent manner while cell lines with low29 (MCF7) or absent30 (CHO-K1) EGFR expression are not transduced.
In this study we describe the first viral system with chemically switchable infectivity. This system perfectly complements previously developed synthetic biological switches for controlling intracellular processes.1e,4 While all these switches focus on transgene systems already installed into the cell, AAVSWITCH allows for the first time the direct chemically switchable control of the gene transfer process itself based on highly efficient viral vectors.31 Beyond the described specific implementation of AAVSWITCH, we assume that the here-presented approach is likely generically applicable to different viral vector systems, target cell lines and chemical inducers by modularly exchanging the dimerizer modules or the cell-binding domain. The concept presented in this study thus represents a blueprint for obtaining a time-resolved control of the cell specificity of viral vectors in order to precisely target molecular interventions in future synthetic biological devices and gene therapy scenarios.
We would like to thank Silke Uhrig, Hanna Janicki and Laura Escalona-Espinosa for excellent technical assistance. This work was supported by the European Research Council under the European Community's Seventh Framework Programme (FP7/2007–2013)/ERC Grant agreement no 259043-CompBioMat, the Initiating and Networking Fund (IVF) of the Helmholtz Association within the Helmholtz Initiative on Synthetic Biology (SO-078) and the German Excellence Initiative of the Federal and State Governments (GSC-4 and EXC-294). GC, EW and DG were supported by the Cluster of Excellence CellNetworks (EXC81). HB was supported by the Center for Molecular Medicine Cologne (2-GB) and the DFG Priority Program 1230 “Mechanisms of gene vector entry and persistence” (BU1310/1-2). Imaging was performed with the kind support of the Life Imaging Center (LIC) in the Center for Systems Biology (ZBSA), University of Freiburg.
Notes and references
- (a) P. E. Purnick and R. Weiss, Nat. Rev. Mol. Cell Biol., 2009, 10, 410 CrossRef CAS PubMed; (b) A. L. Slusarczyk, A. Lin and R. Weiss, Nat. Rev. Genet., 2012, 13, 406 CrossRef CAS PubMed; (c) W. C. Ruder, T. Lu and J. J. Collins, Science, 2011, 333, 1248 CrossRef CAS PubMed; (d) A. L. Chang, J. J. Wolf and C. D. Smolke, Curr. Opin. Biotechnol., 2012, 23, 679 CrossRef CAS PubMed; (e) M. Hörner and W. Weber, FEBS Lett., 2012, 586, 2084 CrossRef PubMed.
- (a) A. Kume, K. Ito, Y. Ueda, M. Hasegawa, M. Urabe, H. Mano and K. Ozawa, Biochem. Biophys. Res. Commun., 1999, 260, 9 CrossRef CAS PubMed; (b) T. Neff, P. A. Horn, V. E. Valli, A. M. Gown, S. Wardwell, B. L. Wood, C. von Kalle, M. Schmidt, L. J. Peterson, J. C. Morris, R. E. Richard, T. Clackson, H. P. Kiem and C. A. Blau, Blood, 2002, 100, 2026 CrossRef CAS PubMed.
- A. V. Karginov, F. Ding, P. Kota, N. V. Dokholyan and K. M. Hahn, Nat. Biotechnol., 2010, 28, 743 CrossRef CAS PubMed.
- S. Ausländer and M. Fussenegger, Trends Biotechnol., 2013, 31, 155 CrossRef PubMed.
- (a) K. M. Bonger, L. C. Chen, C. W. Liu and T. J. Wandless, Nat. Chem. Biol., 2011, 7, 531 CrossRef CAS PubMed; (b) K. Nishimura, T. Fukagawa, H. Takisawa, T. Kakimoto and M. Kanemaki, Nat. Methods, 2009, 6, 917 CrossRef CAS PubMed.
- M. Dühren-von Minden, R. Übelhart, D. Schneider, T. Wossning, M. P. Bach, M. Buchner, D. Hofmann, E. Surova, M. Follo, F. Köhler, H. Wardemann, K. Zirlik, H. Veelken and H. Jumaa, Nature, 2012, 489, 309 CrossRef PubMed.
- W. Weber and M. Fussenegger, Nat. Rev. Genet., 2012, 13, 21 CAS.
- (a) X. Ye, V. M. Rivera, P. Zoltick, F. Cerasoli Jr., M. A. Schnell, G. Gao, J. V. Hughes, M. Gilman and J. M. Wilson, Science, 1999, 283, 88 CrossRef CAS; (b) A. Auricchio, V. M. Rivera, T. Clackson, E. E. O'Connor, A. M. Maguire, M. J. Tolentino, J. Bennett and J. M. Wilson, Mol. Ther., 2002, 6, 238 CrossRef CAS; (c) P. Hadaczek, J. Beyer, A. Kells, W. Narrow, W. Bowers, H. J. Federoff, J. Forsayeth and K. S. Bankiewicz, PLoS One, 2011, 6, e27728 CAS; (d) L. M. Sanftner, V. M. Rivera, B. M. Suzuki, L. Feng, L. Berk, S. Zhou, J. R. Forsayeth, T. Clackson and J. Cunningham, Mol. Ther., 2006, 13, 167 CrossRef CAS PubMed.
- (a) C. S. Hackett, A. M. Geurts and P. B. Hackett, Genome Biol., 2007, 8(suppl 1), S12 CrossRef PubMed; (b) P. Maier, C. von Kalle and S. Laufs, Future Microbiol., 2010, 5, 1507 CrossRef CAS PubMed.
- (a) A. S. Wierzbicki and A. Viljoen, Expert Opin. Biol. Ther., 2013, 13, 7 CrossRef CAS PubMed; (b) M. Nonnenmacher and T. Weber, Gene Ther., 2012, 19, 649 CrossRef CAS PubMed; (c) A. Asokan, J. B. Hamra, L. Govindasamy, M. Agbandje-McKenna and R. J. Samulski, J. Virol., 2006, 80, 8961 CrossRef CAS PubMed.
- S. R. Opie, K. H. Warrington Jr., M. Agbandje-McKenna, S. Zolotukhin and N. Muzyczka, J. Virol., 2003, 77, 6995 CrossRef CAS.
- J. Choi, J. Chen, S. L. Schreiber and J. Clardy, Science, 1996, 273, 239 CAS.
- K. Lux, N. Goerlitz, S. Schlemminger, L. Perabo, D. Goldnau, J. Endell, K. Leike, D. M. Kofler, S. Finke, M. Hallek and H. Buning, J. Virol., 2005, 79, 11776 CrossRef CAS PubMed.
- J. Chen, X. F. Zheng, E. J. Brown and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4947 CrossRef CAS.
- M. Leone, K. J. Crowell, J. Chen, D. Jung, G. G. Chiang, S. Sareth, R. T. Abraham and M. Pellecchia, Biochemistry, 2006, 45, 10294 CrossRef CAS PubMed.
- S. Szep, S. Park, E. T. Boder, G. D. Van Duyne and J. G. Saven, Proteins, 2009, 74, 603 CrossRef CAS PubMed.
- D. Steiner, P. Forrer and A. Plückthun, J. Mol. Biol., 2008, 382, 1211 CrossRef CAS PubMed.
- (a) R. Pollock, M. Giel, K. Linher and T. Clackson, Nat. Biotechnol., 2002, 20, 729 CrossRef CAS PubMed; (b) J. H. Bayle, J. S. Grimley, K. Stankunas, J. E. Gestwicki, T. J. Wandless and G. R. Crabtree, Chem. Biol., 2006, 13, 99 CrossRef CAS PubMed; (c) S. Indraccolo, L. Moserle, V. Tisato, E. Gola, S. Minuzzo, V. Roni, L. Persano, L. Chieco-Bianchi and A. Amadori, Gene Ther., 2006, 13, 953 CrossRef CAS PubMed.
- H. S. Wiley, Exp. Cell Res., 2003, 284, 78 CrossRef CAS.
- X. Xiao, J. Li and R. J. Samulski, J. Virol., 1998, 72, 2224 CAS.
- R. C. Münch, H. Janicki, I. Völker, A. Rasbach, M. Hallek, H. Büning and C. J. Buchholz, Mol. Ther., 2013, 21, 109 CrossRef PubMed.
- D. Grimm, K. L. Streetz, C. L. Jopling, T. A. Storm, K. Pandey, C. R. Davis, P. Marion, F. Salazar and M. A. Kay, Nature, 2006, 441, 537 CrossRef CAS PubMed.
- L. S. Chang and T. Shenk, J. Virol., 1990, 64, 2103 CAS.
- K. N. Leppard, J. Gen. Virol., 1997, 78, 2131 CAS.
- R. Nayak and D. J. Pintel, J. Virol., 2007, 81, 11908 CrossRef CAS PubMed.
- (a) D. Grimm, J. S. Lee, L. Wang, T. Desai, B. Akache, T. A. Storm and M. A. Kay, J. Virol., 2008, 82, 5887 CrossRef CAS PubMed; (b) D. Grimm, S. Zhou, H. Nakai, C. E. Thomas, T. A. Storm, S. Fuess, T. Matsushita, J. Allen, R. Surosky, M. Lochrie, L. Meuse, A. McClelland, P. Colosi and M. A. Kay, Blood, 2003, 102, 2412 CrossRef CAS PubMed.
- M. L. Janmaat, F. A. Kruyt, J. A. Rodriguez and G. Giaccone, Clin. Cancer Res., 2003, 9, 2316 CAS.
- F. M. Sirotnak, M. F. Zakowski, V. A. Miller, H. I. Scher and M. G. Kris, Clin. Cancer Res., 2000, 6, 4885 CAS.
- C. Mamot, D. C. Drummond, U. Greiser, K. Hong, D. B. Kirpotin, J. D. Marks and J. W. Park, Cancer Res., 2003, 63, 3154 Search PubMed.
- S. Goldoni, R. A. Iozzo, P. Kay, S. Campbell, A. McQuillan, C. Agnew, J. X. Zhu, D. R. Keene, C. C. Reed and R. V. Iozzo, Oncogene, 2007, 26, 368 CrossRef CAS PubMed.
- F. Mingozzi and K. A. High, Nat. Rev. Genet., 2011, 12, 341 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6, Tables S1 and S2, and Materials and methods. See DOI: 10.1039/c4cc03292f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |