 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjugated microporous polytriphenylamine networks†
Yaozu
Liao
a,
Jens
Weber
b and
Charl F. J.
Faul
*a
aSchool of Chemistry, University of Bristol, Bristol, England BS8 1TS, UK. E-mail: charl.faul@bristol.ac.uk
bHochschule Zittau/Görlitz (University of Applied Science), Department of Chemistry, Theodor-Körner-Allee 16, D-02763 Zittau, Germany
First published on 4th June 2014
Abstract
Conjugated microporous polytriphenylamine networks with surface areas of 530 m2 g−1 were synthesized via Buchwald–Hartwig coupling, resulting in high CO2 uptake (up to 6.5 wt%) and CO2–N2 selectivity (75) at 1 bar and 303 K.
Conjugated microporous polymers (CMPs) with intrinsic properties including small pore sizes (<2 nm), large specific surface areas (>6000 m2 g−1),1 high chemical stability, low skeleton density and reversible redox properties exhibit great promise for gas storage, separation, sensing, catalysis and battery applications.2 Versatile CMPs have been obtained readily through template-free chemical processes by careful selection of building blocks and, commonly, suitable C–C or C–N coupling reactions, which showed efficient preparation and high flexibility in the molecular design. Typically, Suzuki,3 Sonogashira–Hagihara4 and Yamamoto coupling,5 Schiff-base chemistry,6 cyclotrimerization7 and oxidative polymerization8 reactions have been used to synthesize CMPs. Previous studies demonstrated that control over average micropore size, surface area, and gas uptake of the CMPs was enabled by alternating the strut lengths, rigidities, and functionalities of the building blocks.2
Buchwald–Hartwig (BH) coupling is utilized in organic synthesis for the generation of C–N bonds via the palladium-catalysed cross-coupling of amines with aryl halides. The BH coupling approach allows for expansion of the repertoire of possible C–N bond formation through the facile synthesis of aromatic amines. This useful method also provides a simple route to nitrogen-containing redox-active systems, as shown in recent work9 from our laboratories on the synthesis of well-defined symmetrical oligo(aniline)s, OANIs. It is noteworthy that BH coupling has rarely been used for CMP preparation.10
Here we report the synthesis of conjugated microporous polytriphenylamine (PTPA) networks with a surface area up to 530 m2 g−1 using BH coupling (Scheme 1). Our target was to design and synthesise intrinsically microporous materials based on conjugated polymer networks from a triphenylamine bromide core and selected aryl amine linkers. This approach, exploiting careful choice of strut lengths, rigidities of the linkers and additional included functionalities, would allow formation of nitrogen-containing CMPs with fine-tuned porosities and CO2 adsorption/separation capabilities.
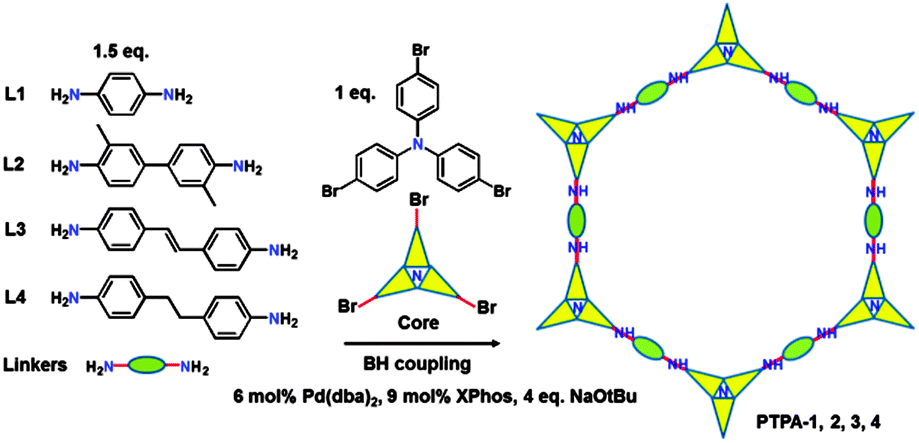 | ||
| Scheme 1 Synthetic route to conjugated microporous PTPAs. | ||
Verification of this approach was achieved by BH cross-coupling of aniline with tris(4-bromophenyl)amine. This resulted in C–N bond formation, affording the fully reduced molecular analogue of the PTPAs, 4,4′,4′′-tris(phenylamino)triphenylamine (analogous to the leucoemeraldine base structure found for OANIs, see ESI,† Scheme S1). Once verified, BH coupling of diamine species afforded black, blue, brown, and yellow insoluble PTPA-1, 2, 3, 4 powders (Fig. S1, ESI†) with yields of 65–80%. The chemical structures of the PTPAs were confirmed by Fourier transform infrared (FT-IR) and solid-state 13C cross-polarization magic angle spinning nuclear magnetic resonance (CP/MAS NMR) investigations. Bands of the primary amine group of the linkers at 3470 and 3420 (–NH2 stretching) and 1650 cm−1 (–NH2 deformation) as well as the aromatic C–Br groups of the tris(4-bromophenylamine) at 1178 cm−1 (aromatic C–Br stretching) are absent or strongly attenuated in the spectra of the PTPAs (Fig. S2, ESI†). The distinct quinoid (Q) and benzenoid (B) bands at 1598 and 1498 cm−1 as well as the aryl C–H band at 820 cm−1 are present in the spectra of the resulting materials. Solid-state 13C CP/MAS NMR spectra of all polymers showed three main resonances at ∼141, ∼127 and ∼118 ppm, originating from the aryl carbons of both the starting materials (Fig. S3, ESI†). Three additional resonances at 17, 116 and 35 ppm are attributed to the methyl, alkenyl and methylene groups of PTPA-2, 3 and 4, respectively. The ultra-violet/visible near infrared (UV/Vis-NIR) spectra of fully conjugated PTPA-1, PTPA-2, and PTPA-3 exhibited two peaks at 380 and 600 nm, typically attributed to the π–π* transition of benzenoid and quinoid rings (Fig. 1), respectively, as confirmed by recent calculations.9 Specifically, PTPA-3 displayed a broad peak around 1100 nm, attributed to the electron and hole migration found in trans-stilbene.11 These results confirm the creation of extended conjugated PTPA networks with amine linkages.
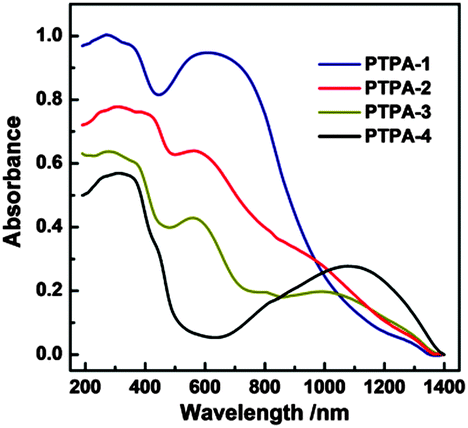 | ||
| Fig. 1 Solid-state UV/Vis-NIR spectra of PTPAs. | ||
As expected, PTPAs displayed amorphous structures, as determined by powder X-ray diffraction (XRD) measurements (broad peaks at 2θ = ∼12.2°, Fig. S4, ESI†). No π-stacking was evident from these investigations. Scanning electron microscope (SEM) images showed the morphologies of PTPAs consisting of aggregated nanoparticles with diameters of 200–500 nm (Fig. 2). This leads to some outer surface area (large meso- and macropores due to interstitial voids), which is highest for PTPA-2 and PTPA-3 as evidenced by cryogenic gas adsorption (see below). The transmission electron microscope (TEM) images indicate the microporous structures of the PTPA nanoparticles (see Fig. S5, ESI†), which are typically found in amorphous CMPs. The materials are thermally stable in N2 up to 500 °C, as revealed by thermal gravimetric analysis (TGA) (see Fig. S6, ESI†). In particular, for PTPA-3 and PTPA-4, ∼60 wt% remained after heating to 1000 °C. The excellent thermal stability of PTPAs is ascribed to the nature of their cross-linked networks.
 | ||
| Fig. 2 SEM images of prepared PTPAs. | ||
The porosity parameters of the polymers were initially studied by gas adsorption analysis using nitrogen as the adsorbate. Nitrogen adsorption–desorption isotherms of PTPAs measured at 77.4 K are shown in Fig. 3. All materials showed some outer surface area, most likely originating from interparticulate porosity associated with the meso- and macrostructures of the samples and interparticular voids. This was evidenced by the steep increase of N2 uptake at relative pressure above 0.9 observed for all materials. It is plausible to expect that precipitation of the growing polymer networks is responsible for the formation of observed macro/mesoporosity.
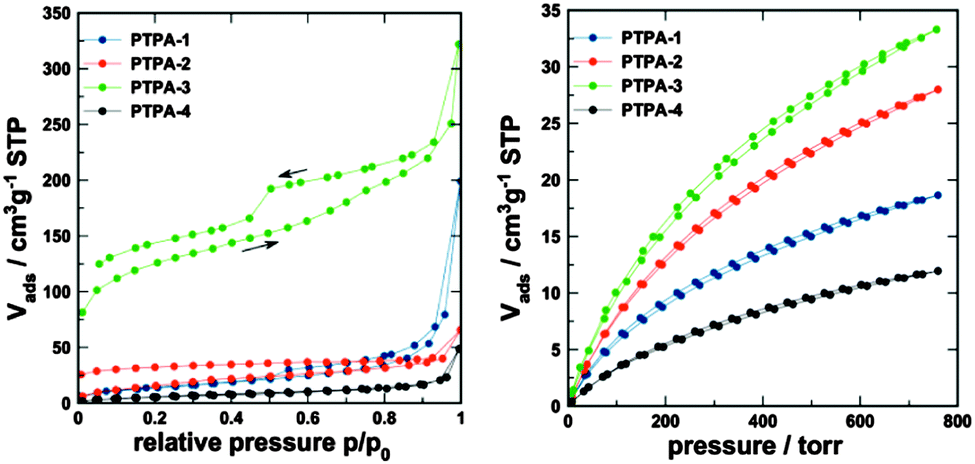 | ||
| Fig. 3 (left) N2 adsorption–desorption isotherms (77.4 K) and (right) CO2 adsorption–desorption isotherm at 273 K. | ||
PTPA-4 does not show any other signs of porosity and consequently shows the lowest N2 uptake. In contrast, PTPA-1 does show some broadly distributed mesoporosity in conjunction with macroporosity, as derived from the hysteresis between the adsorption and desorption curves, which close around p/p0 ∼ 0.45. PTPA-2 and PTPA-3 show a low-pressure hysteresis (the isotherm curves do not close even at very low p/p0), which is often observed for microporous polymers.12 The hysteresis could result from restricted access of the adsorbate to the some of the micropores of the material, especially to those that are blocked by narrow openings. It has been suggested that additional methods such as CO2 adsorption should be used for micropore analysis (see below).13PTPA-3 shows the highest apparent Brunauer–Emmett–Teller (BET) surface areas (adsorption branch: 450 m2 g−1; desorption branch: 530 m2 g−1, see Table 1). Further studies analysing the gas separation potential of this material in more detail is currently under way. PTPA-3 seems to also feature some ill-defined mesoporosity, indicated by the step of the desorption isotherm at the characteristic p/p0 of 0.45 (emptying of ink-bottle mesopores by cavitation). PTPA-2, in contrast, does not show signs of mesoporosity based on N2 analysis. The specific pore volumes and surface areas of the PTPA materials are moderate in comparison to other CMPs. However, the combination of micro-, meso- and macroporosity (especially for the PTPA-3 with a determined pore volume of 0.36 cm3 g−1) is of interest for applications that require good mass transfer through so-called transport pores.
| PTPA | S BET (m2 g−1) | PVc (cm3 g−1) | MPVd (cm3 g−1) | μ-pore SGCMCe (m2 g−1) | MPVGCMCe (cm3 g−1) | CO2 uptake at 1.0 bar (wt%) | |
|---|---|---|---|---|---|---|---|
| 273 K | 303 K | ||||||
| a Surface area calculated from nitrogen adsorption isotherms using the BET equation. b Surface area calculated from the desorption branch of the N2 isotherms using the BET equation. c Pore volume calculated from nitrogen adsorption at p/p0 = 0.95. d Micropore volume calculated from QSDFT (slit pore model, desorption branch) analysis of N2 data. e Micropore surface area determined from GCMC analysis of the CO2 adsorption data at 273 K. n.a. not applicable. n.d. not determined. | |||||||
| 1 | 52 | 0.11 | n.a. | 195 | 0.07 | 3.7 | n.d. |
| 2 | 62/120b | 0.06 | 0.05 | 292 | 0.11 | 5.5 | 2.6 |
| 3 | 450/530b | 0.36 | 0.18 | 356 | 0.13 | 6.5 | 3.4 |
| 4 | 22 | 0.03 | n.a. | 130 | 0.05 | 2.2 | n.d. |
CO2 adsorption was measured for all PTPA materials at 273 K (see Fig. 3) to gain further information on the microporosity.14 All materials showed significant CO2 uptake. The highest uptake was found for PTPA-3 (6.5 wt%), followed by PTPA-2 (5.5 wt%), and PTPA-1 and PTPA-4 (uptake <4 wt%). The isotherms showed only weak hysteresis and were analysed using the commercialized Grand-canonical Monte-Carlo (GCMC), methodology to extract porosity information (see Table 1).15 All materials show similar broad pore size distributions, which is in line with their amorphous nature. The main fraction of pores have sizes between 0.5 and 1 nm (Fig. S7, ESI†).
Generally, PTPAs prepared from monomers containing longer and more rigid struts should show higher apparent specific surface areas if no interdigitating structures are observed. Shorter linkers might lead to compact structures of low porosity. However, the monomer geometry alone is not decisive in determining final porosity, as the reaction conditions, choice of solvents and solubility of the forming polymeric network phase all contribute to the physical properties of the final CMPs. It is nevertheless of worth to discuss the observed porosities with regard to the monomer geometry and rigidity, allowing comparison and conclusions to be drawn for the present systems. The use of the stilbene monomer (L3), which is significantly more rigid compared with its ethylene-bridged counterpart L4, results in the formation of significant microporosity. Interestingly, the very rigid (but shorter) p-phenylenediamine (L1) yields a moderately microporous material. The low microporosity could result from the formation of a more dense structure (PTPA-1), i.e. the conjugated segments are less separated and might adhere to each other, favouring pore closure. The methyl-decorated biphenylene-based linker L2 yields a network of considerable microporosity. The strategy to enhance the free volume of, for example, membrane polymers using such methyl-decorated monomers is well known,16 and is effective here as well. In the investigated systems monomer molecular structures do impact on the observed porosity, thus following commonly known trends. It also shows that our chosen platform is modular and that porosities could be tuned to reach higher values through further exploration and optimization of the reaction conditions.
The presence of fairly small pores with a uniform distribution of polar amine units is expected to result in favourable interactions between specific adsorbate molecules and the polymeric adsorbent. We analysed the potential of PTPA-3 for gas separation, exemplified by the CO2–N2 gas pair. The separation of CO2 from, for example, flue gas (post-combustion) is of current interest with regard to the capture of CO2 for environmental (greenhouse gas) or economic reasons (CO2 as alternative feedstock for new polymers). There are various other applications, such as cryogenic spray-freezing and food preservation that would benefit from CO2 capture. CO2 adsorption on PTPA-3 at 30 °C (303 K) was 0.765 mmol g−1 (3.4 wt% at 1.0 bar), lower compared with some standard materials (zeolites, activated carbon). The N2 adsorption on PTPA-3 at 30 °C was found to be 0.022 mmol g−1 at 1.0 bar, resulting in an equilibrium selectivity of α(CO2/N2)eq ∼ 35. Further analysis was performed by calculating the IAST selectivity at 30 °C based on the assumption of a gas composition of 15% (v/v) CO2 balanced with N2. The necessary description of the isotherm was achieved by fitting the experimental data points by either a simple Langmuir (N2) or a dual-site Langmuir fit (CO2). It should however be noted that the CO2 data could also be described very well by a simple Langmuir fit (see Fig. S8, ESI†). The IAST selectivity of CO2 over N2 is calculated to be 75 at ambient pressure (1.0 bar, 30 °C). This value compares very favourably with most reported results for microporous organic polymers, usually with selectivities in the range 20 to 50.17 The predicted adsorption isotherms from the gas mixture are shown in Fig. 4.
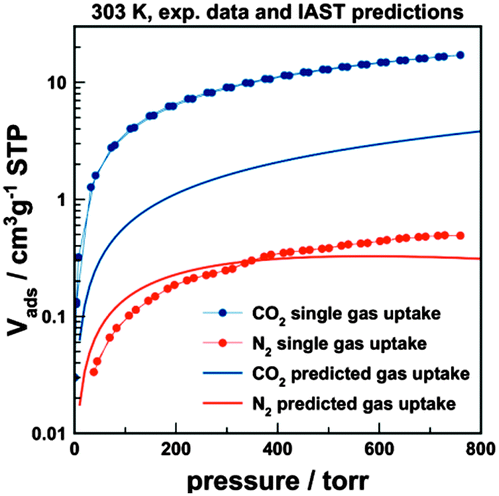 | ||
| Fig. 4 CO2 and N2 adsorption–desorption isotherms (single gas) for PTPA-3 at 303 K (symbol + line) with predicted adsorption isotherms (line only) from a 0.15/0.85 v/v gas mixture of CO2 and N2 at 303 K (based on IAST methodology). | ||
The fact that the CO2 adsorption isotherm could also be represented by a simple Langmuir approach indicates that the surface area of PTPA-3 is very homogenous. This is indeed supported by first estimation of the isosteric heat of adsorption qst based on the CO2 isotherms measured at 273 and 303 K. The qst remained almost constant over the whole coverage range (values between 27 and 28 kJ mol−1) indicating a very stable operation regime. The qst values are somewhat higher than values commonly found for activated carbons or other polymers, but low enough for adsorbent regeneration without too high an energetic penalty.
In summary, we have successfully synthesized a series of conjugated microporous polytriphenylamine (PTPAs) based on a triphenylamine core motif using Buchwald–Hartwig (BH) cross-coupling. The BET surface area of these conjugated microporous polymers is up to 530 m2 g−1. Owing to the microporosity and the homogenously electron-rich and polar pore surface, they exhibit high CO2 uptakes of up to 6.5 wt% and good CO2–N2 adsorption ideal selectivities of up to 75 at 303 K and 1.0 bar. The overall porosity and the corresponding gas adsorption properties strongly depend on the building block size, molecular architectures and functionalities. BH coupling presents a promising approach for large-scale manufacturing of conjugated microporous polymers and provides a high conversion of functional monomers used. Post-synthesis treatment of the tertiary and secondary amines resulting in permanent quarterisation and careful choice of corresponding counterions may enable tuning of gas uptake and selectivity. Continued investigation into the design, synthesis and characterisation of related materials is on-going in our laboratories.
We are grateful to the European Commission Marie Curie International Incoming Fellowship (FP7-PEOPLE-2012-IIF TANOGAPPs No. 326385) for generous support of this project.
Notes and references
- D. Q. Yuan, W. G. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723 CrossRef CAS PubMed.
- (a) J.-X. Jiang, A. Trewin, F. B. Su, C. D. Wood, H. J. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658 CrossRef CAS; (b) F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819 RSC; (c) Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- (a) J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334 CrossRef CAS PubMed; (b) L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742 CrossRef CAS PubMed; (c) D. B. Xiao, Y. Li, L. L. Liu, B. Wen, Z. J. Gu, C. Zhang and Y. S. Zhao, Chem. Commun., 2012, 48, 9519 RSC; (d) Y. H. Xu, A. Nagai and D. L. Jiang, Chem. Commun., 2013, 49, 1591 RSC.
- (a) J. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed; (b) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS PubMed; (c) R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809 CrossRef CAS; (d) Q. Chen, M. Luo, T. Wang, J.-X. Wang, D. Zhou, Y. Han, C.-S. Zhang, C.-G. Yan and B.-H. Han, Macromolecules, 2011, 44, 5573 CrossRef CAS.
- (a) J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426 CrossRef CAS; (b) K. Zhang, B. Tieke, J. C. Forgie and P. J. Skabara, Macromol. Rapid Commun., 2009, 30, 1834 CrossRef CAS PubMed.
- (a) M. G. Schw, B. Fassbender, H. W. Spiess, A. Thomas, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 7216 CrossRef PubMed; (b) G. Y. Li, B. Zhang, J. Yan and Z. G. Wang, Chem. Commun., 2014, 50, 1897 RSC.
- (a) R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., 2009, 121, 7042 CrossRef; (b) S. J. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357 CrossRef CAS PubMed; (c) X. Zhu, C. Tian, S. M. Mahurin, S. H. Chai, C. M. Wang, S. Brown, G. M. Veith, H. M. Luo, H. L. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478 CrossRef CAS PubMed.
- (a) J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702 CrossRef CAS; (b) Y. L. Luo, B. Y. Li, W. Wang, K. B. Wu and B. E. Tan, Adv. Mater., 2012, 24, 5703 CrossRef CAS PubMed.
- Z. C. Shao, P. Rannou, S. Sadki, N. Fey, D. M. Lindsay and C. F. J. Faul, Chem. – Eur. J., 2011, 17, 12512 CrossRef CAS PubMed.
- J. Germain, F. Svec and J. M. J. Fréchet, Chem. Mater., 2008, 20, 7069 CrossRef CAS.
- A. Moissett, R. F. Lobo, H. Vezin, K. A. Al-Majnouni and C. Brémard, J. Phys. Chem. C, 2010, 114, 10280 Search PubMed.
- P. Kuhn, A. Forget, D. S. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333 CrossRef CAS PubMed.
- J. Jeromenok and J. Weber, Langmuir, 2013, 29, 12982 CrossRef CAS PubMed.
- (a) D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233 CrossRef PubMed; (b) N. Ritter, I. Senkovska, S. Kaskel and J. Weber, Macromolecules, 2011, 44, 2025 CrossRef CAS.
- P. I. Ravikovitch, A. Vishnyakov, R. Russo and A. V. Neimark, Langmuir, 2000, 16, 2311 CrossRef CAS.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907 CrossRef CAS.
- (a) R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 RSC; (b) Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Studies on the synthesis, characterization, morphology, and gas adsorption–desorption of the PTPAs. See DOI: 10.1039/c4cc03026e |
| This journal is © The Royal Society of Chemistry 2014 |