 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of nitrogen heterocycles via α-aminoalkyl radicals generated from α-silyl secondary amines under visible light irradiation†
Kazunari
Nakajima
,
Mai
Kitagawa
,
Yuya
Ashida
,
Yoshihiro
Miyake‡
* and
Yoshiaki
Nishibayashi
*
Institute of Engineering Innovation, School of Engineering, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: ynishiba@sogo.t.u-tokyo.ac.jp
First published on 16th June 2014
Abstract
We have succeeded in the visible light-mediated synthetic use of α-aminoalkyl radicals derived from α-silyl secondary amines toward addition to α,β-unsaturated carbonyl compounds. The resulting γ-aminocarbonyl compounds are converted into γ-lactams and pyrroles in a one-pot process.
Five-membered nitrogen heterocycles such as γ-lactams and pyrroles are found in various natural products and pharmaceuticals.1,2 Construction of these skeletons in a simple and efficient manner is one of the most important topics in synthetic organic chemistry. In this context, the development of a novel synthetic approach for γ-aminocarbonyl compounds composed of a secondary or primary amine moiety plays a pivotal role because a rapid 5-exo-cyclization between amine and carbonyl groups gives the corresponding nitrogen heterocycles.1
Construction of γ-aminocarbonyl skeletons by the addition of α-aminoalkyl radicals derived from secondary amines to α,β-unsaturated carbonyl compounds under the photoinduced electron transfer conditions is a potentially useful strategy to access the corresponding nitrogen heterocycles.3 However, in these reaction systems, a large amount of N-alkylation products was usually observed due to the formation of aminyl radicals by oxidation of secondary amines (Scheme 1a)4,5 or direct thermal aza-Michael addition,6 where the desired γ-aminocarbonyl compounds were obtained only in low yields.3 As a result, successful examples for synthetic utilization of α-aminoalkyl radicals under UV3,7 or visible light8,9 irradiation have been strictly limited to the use of tertiary amine derivatives. These results indicate that the generation and utilization of α-aminoalkyl radicals derived from secondary amines are quite difficult.
 | ||
| Scheme 1 Photoreactions of secondary amines via (a) aminyl radicals and (b) α-aminoalkyl radicals. | ||
Hence, we have envisaged the use of α-silylamines as substrates because dissociation of the α-C–Si bond of the radical cations occurs readily, which is considered to be suitable for selective formation of α-aminoalkyl radicals over aminyl radicals (Scheme 1b).10,11 In fact, we have recently succeeded in the synthetic utilization of α-aminoalkyl radicals generated from α-silyl tertiary amines in the presence of visible light-photoredox catalysts.8,12 In the course of our study, we have disclosed visible light-mediated synthetic utilization of α-aminoalkyl radicals generated from α-silyl secondary amines toward the addition to α,β-unsaturated carbonyl compounds, where the produced γ-aminocarbonyl compounds are converted into the corresponding γ-lactams and pyrroles in simple one-pot procedures. The preliminary results are described here.
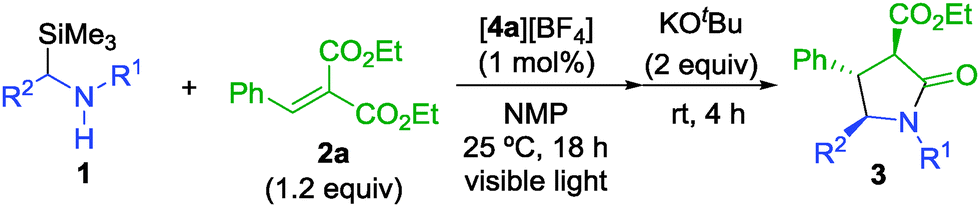
Initial investigations of one-pot synthesis of γ-lactams were conducted using N-(trimethylsilylmethyl)aniline (1a) and diethyl benzylidenemalonate (2a) as substrates (Table 1). In the presence of 1 mol% of [4a][BF4], the reaction of 1a with 1.2 equiv. of 2a was carried out in N-methylpyrrolidone (NMP) under visible light illumination using a 14 W white LED at 25 °C for 18 h. After the photoreaction, treatment of the resulting reaction mixture with 2 equiv. of KOtBu afforded N-phenyl-3-ethoxycarbonyl-4-phenyl-2-pyrrolidone (3a) in 89% yield (Table 1, entry 1). In the absence of base, uncyclized product (3a′) was obtained in 58% yield along with 3a in 34% yield (Table 1, entry 2). These results indicate that addition of base is important to obtain the γ-lactam in a high yield. When reactions were carried out in other polar solvents such as N,N-dimethylformamide (DMF) and dimethylsulfoxide (DMSO), 3a was obtained in lower yields (Table 1, entries 3 and 4). Ethanol and tetrahydrofuran (THF) were not effective solvents in this reaction system (Table 1, entries 5 and 6). Use of other iridium complexes ([4b][BF4] and [4c][BF4]) and ruthenium(II) tris(2,2′-bipyridyl) complex ([Ru(bpy)3][BF4]2) as photocatalysts gave 3a in slightly lower yields (Table 1, entries 7–9). Separately, we confirmed that the photoreaction did not proceed at all in the absence of photocatalysts or visible light illumination.
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Solvent | Yield of 3ab (%) | Trans/cisc |
| a All reactions of 1a (0.25 mmol) with 2a (0.30 mmol) were carried out in the presence of a photocatalyst (0.0025 mmol) in solvent (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. Then, the resulting mixture was treated with KOtBu (0.50 mmol) at room temperature for 4 h. b Isolated yield. c The isomeric ratio was determined by 1H NMR of the crude products. d The isomeric ratio of the isolated 3a is 14/1. e Without treatment with KOtBu. f 3a′ is obtained in 58% yield. | ||||
| 1 | [4a][BF4] | NMP | 89 | >20/1d |
| 2e,f | [4a][BF4] | NMP | 34 | >20/1 |
| 3 | [4a][BF4] | DMF | 83 | >20/1 |
| 4 | [4a][BF4] | DMSO | 73 | 12/1 |
| 5 | [4a][BF4] | EtOH | 31 | 14/1 |
| 6 | [4a][BF4] | THF | 6 | >20/1 |
| 7 | [4b][BF4] | NMP | 74 | >20/1 |
| 8 | [4c][BF4] | NMP | 77 | >20/1 |
| 9 | [Ru(bpy)3][BF4]2 | NMP | 86 | >20/1 |

|
||||

Next, we investigated reactions of various α-silylamines (1) with 2a (Table 2). Use of α-silylamines bearing an electron-withdrawing or donating group on the benzene ring afforded the corresponding products in excellent yields (Table 2, entries 1–5). Introduction of a naphthyl group instead of a phenyl ring was successful to give 3g in 75% yield (Table 2, entry 6). Dialkylamine derivative 1h was also applicable to this reaction system to give the corresponding products in 44% yields (Table 2, entry 7). The use of an α-aminoethyl radical generated from 1-(trimethylsilyl)ethyl amine (1i) gave the corresponding tri-substituted γ-lactam (3i) in 62% yield (Table 2, entry 8). Unfortunately, when a primary amine derivative (trimethylsilylmethyl)amine and an amide derivative N-(trimethylsilylmethyl)acetamide were used as substrates, no formation of the corresponding products was observed.
|
|
|||
|---|---|---|---|
| Entry | α-Silylamine (1) | Yield of 3b (%) | Trans/cisc |
| a All reactions of 1 (0.25 mmol) with 2a (0.30 mmol) were carried out in the presence of [4a][BF4] (0.0025 mmol) in NMP (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. Then, the resulting mixture was treated with KOtBu (0.50 mmol) at room temperature for 4 h. b Isolated yield. c The isomeric ratio was determined by 1H NMR of the isolated products. d Treatment with KOtBu was done at 100 °C for 17 h. e For 48 h of photoreaction. f 1h (0.30 mmol) and 2a (0.25 mmol) were used. g The ratio of two major isomers. | |||
| 1 | R1 = 4-CIC6H4, R2 = H (1b) | 95 (3b) | >20/1 |
| 2 | R1 = 4-FC6H4, R2 = H (1c) | 85 (3c) | 19/1 |
| 3 | R1 = 4-MeC6H4, R2 = H (1d) | 90 (3d) | 20/1 |
| 4d | R1 = 2-MeC6H4, R2 = H (1e) | 66 (3e) | 15/1 |
| 5e | R1 = 4-MeOC6H4, R2 = H (1f) | 79 (3f) | 6/1 |
| 6 | R1 = 1-Naphthyl, R2 = H (1g) | 75 (3g) | 9/1 |
| 7d,f | R1 = tBu, R2 = H (1h) | 44 (3h) | 7/1 |
| 8 | R1 = Ph, R2 = Me (1i) | 62 (3i) | 6/1g |
Reactions of N-(trimethylsilylmethyl)aniline (1a) with a variety of α,β-unsaturated esters (2) were examined (Table 3). Use of α,β-unsaturated esters bearing various aromatic and alkyl groups gave the corresponding products in high yields (Table 3, entries 1–7). Introduction of a cyano group instead of an ester group was successful to give 3q in 81% yield (Table 3, entry 8). When ethyl acrylate (2j) was used, 3r was obtained in 47% yield (Table 3, entry 9). Tetra-substituted alkene 2k was also applicable to give the corresponding γ-lactam (3s) in 77% yield (Table 3, entry 10).
|
|
|||
|---|---|---|---|
| Entry | α,β-Unsaturated ester (2) | Yield of 3b (%) | Trans/cisc |
| a All reactions of 1a (0.25 mmol) with 2 (0.30 mmol) were carried out in the presence of [4a][BF4] (0.0025 mmol) in NMP (2.5 mL) under 14 W white LED illumination at 25 °C for 18 h. Then, the resulting mixture was treated with KOtBu (0.50 mmol) at room temperature for 4 h. b Isolated yield. c The isomeric ratio was determined by 1H NMR of the isolated products. d Treatment with KOtBu was done at 100 °C for 17 h. | |||
| 1 | R1 = 4-CIC6H4, R2 = H, R3 = CO2Et (2b) | 79 (3j) | >20/1 |
| 2 | R1 = 4-MeC6H4, R2 = H, R3 = CO2Et (2c) | 79 (3k) | 20/1 |
| 3 | R1 = 3-MeC6H4, R2 = H, R3 = CO2Et (2d) | 62 (31) | >20/1 |
| 4 | R1 = 4-MeOC6H4, R2 = H, R3 = CO2Et (2e) | 81 (3m) | 20/1 |
| 5 | R1 = 4-PhC6H4, R2 = H, R3 = CO2Et (2f) | 61 (3n) | 14/1 |
| 6 | R1 = 2-Naphthyl, R2 = H, R3 = CO2Et (2g) | 61 (3o) | 19/1 |
| 7 | R1 = nPr, R2 = H, R3 = CO2Et (2h) | 79 (3p) | 11/1 |
| 8d | R1 = Ph, R2 = H, R3 = CN (2i) | 81 (3q) | 8/1 |
| 9 | R1 = R2 = R3 = H (2j) | 47 (3r) | — |
| 10 | R1 = R2 = Me, R3 = CO2Et (2k) | 77 (3s) | — |

The success in synthesis of γ-lactams prompted us to investigate other nitrogen heterocycles such as pyrroles. We have designed α,β-unsaturated ketones as substrates for the construction of a pyrrole ring. When the reaction of 1a with an α,β-unsaturated ketone derivative (5) was examined under similar reaction conditions to the γ-lactam synthesis, dihydropyrrole (6) was obtained in 64% yield (Scheme 2a). Encouraged by this result, we have successfully developed one-pot synthesis of pyrroles by oxidation of dihydropyrroles (Scheme 2b). After photoreactions of α-silyl secondary amines (1a and 1i) with 5, subsequent treatment of the resulting mixture with 2 equiv. of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) at room temperature for 30 min gave the corresponding tri- and tetra-substituted pyrroles (7a and 7b) in high yields.
 | ||
| Scheme 2 One-pot syntheses of dihydropyrrole (6) and pyrroles (7a and 7b). | ||
To obtain mechanistic insight, some additional experiments were carried out. At first, the quantum yield of the reaction of 1a with 2a was determined to be 0.13. The value is in the common range of the photoredox reactions which proceed by a sequential redox process.8 Next, we monitored the photoreaction of 1a with 2a by GC-MS because isolation of the primary products without an aqueous work-up was not possible due to the high boiling point of NMP. GC-MS analyses indicate that the reaction mixture includes hexamethyldisiloxane and 3a′ (see Table 1 for the structure of 3a′). This result shows that the trimethylsilyl group was captured by adventitious water8,10 and 3a′ is the primary product in the reaction system. Separately, we carried out the reaction of 1a with 2a in the presence of a small amount of water (NMP/H2O = 25/1), where 3a was obtained in 86% yield. This result indicates that the additional water did not affect the yield of γ-lactam.
Based on the experimental results, the reactions are considered to proceed via a reaction pathway similar to the previously reported sequential redox pathway, as shown in Scheme 3.8 At first, single electron oxidation of α-silyl secondary amines 1 by a photo-excited catalyst (*cat) occurs. Then, α-aminoalkyl radicals (A) are formed along with generation of trimethylsilyl cations.10 The trimethylsilyl cation is captured by adventitious water in the reaction system to give hexamethyldisiloxane and protons. Addition of A to α,β-unsaturated carbonyl compounds 2 affords the corresponding radical intermediates (B). The reduction of B13 by a reduced catalyst (cat−)14 and subsequent protonation give γ-aminocarbonyl compounds (C) as primary products. Subsequently, base-mediated cyclization of C (E = CO2Et) affords γ-lactams, while oxidation of dihydropyrroles (D) formed by dehydration condensation of C (E = COCH3) gives pyrroles.
 | ||
| Scheme 3 Plausible reaction pathways. | ||
In summary, we have developed a novel reaction system for generation and utilization of α-aminoalkyl radicals derived from secondary amines. The α-aminoalkyl radicals were successfully applied toward addition to α,β-unsaturated carbonyl compounds and subsequent cyclization into nitrogen heterocycles such as γ-lactams and pyrroles. We believe that the method described here provides a useful approach for syntheses of various nitrogen heterocycles, which are useful in pharmacological science. Further investigations on scope of substrates and mechanistic details are now under way.
We thank the Funding Program for Next Generation World-Leading Researchers (GR025) and Grants-in-Aid for Scientific Research (Nos. 26288044, 26620075, 26105708, and 26870120) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Notes and references
- (a) M. B. Smith, in Science of Synthesis, ed. S. M. Weinreb, Theme, Stuttgart, 2005, p. 647 Search PubMed; (b) D. StC. Black, in Science of Synthesis, ed. G. Maas, Theme, Stuttgart, 2001, p. 441 Search PubMed.
- (a) B. Nay, N. Riache and L. Evanno, Nat. Prod. Rep., 2009, 26, 1044 RSC; (b) A. Lebedev, Chem. Heterocycl. Compd., 2007, 43, 673 CrossRef CAS PubMed; (c) H. Hoffmann and T. Lindel, Synthesis, 2003, 1753 CAS; (d) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- (a) S. Das, J. S. D. Kumar, K. Shivaramayya and M. V. George, J. Photochem. Photobiol., A, 1996, 97, 139 CrossRef; (b) S. Das, J. S. D. Kumar, K. Shivaramayya and M. V. George, J. Chem. Soc., Perkin Trans. 1, 1995, 1797 RSC; (c) R. C. Cookson, S. M. deB. Costa and J. Hudec, Chem. Commun., 1969, 753 RSC.
- (a) M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550 CrossRef; (b) F. D. Lewis and P. E. Correa, J. Am. Chem. Soc., 1984, 106, 194 CrossRef CAS; (c) F. D. Lewis, B. E. Zebrowski and P. E. Correa, J. Am. Chem. Soc., 1984, 106, 187 CrossRef CAS; (d) R. W. Fessenden and P. Neta, J. Phys. Chem., 1972, 76, 2857 CrossRef CAS.
- A. G. Fallis and I. M. Brinza, Tetrahedron, 1997, 53, 17543 CrossRef CAS.
- (a) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917 CrossRef CAS; (b) D. Enders, C. Wang and J. X. Liebich, Chem. – Eur. J., 2009, 15, 11058 CrossRef CAS PubMed.
- (a) D. Harakat, J. Pesch, S. Marinković and N. Hoffmann, Org. Biomol. Chem., 2006, 4, 1202 RSC; (b) A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139 CrossRef CAS PubMed; (c) E. S. de Alvarenga, C. J. Cardin and J. Mann, Tetrahedron, 1997, 53, 1457 CrossRef CAS.
- (a) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. – Eur. J., 2014, 20, 6120 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. – Eur. J., 2012, 18, 16473 CrossRef CAS PubMed; (c) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966 RSC; (d) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed.
- (a) H. Zhou, P. Lu, X. Gu and P. Li, Org. Lett., 2013, 15, 5646 CrossRef CAS PubMed; (b) L. R. Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107 CrossRef PubMed; (c) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed; (d) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561 CrossRef CAS; (e) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed; (f) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed.
- (a) D. W. Cho, U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2011, 44, 204 CrossRef CAS PubMed; (b) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233 CrossRef CAS.
- (a) D. Lenhart and T. Bach, Beilstein J. Org. Chem., 2014, 10, 890 CrossRef PubMed; (b) E. Meggers, E. Steckhan and S. Blechert, Angew. Chem., Int. Ed. Engl., 1995, 34, 2137 CrossRef CAS; (c) G. Pandey, G. D. Reddy and G. Kumaraswamy, Tetrahedron, 1994, 50, 8185 CrossRef CAS; (d) G. Pandey, G. Kumaraswamy and U. T. Bhalerao, Tetrahedron Lett., 1989, 30, 6059 CrossRef CAS.
- (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) T. Koike and M. Akita, Synlett, 2013, 2492 CrossRef CAS PubMed; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- J. M. Kern and P. Federlin, Tetrahedron Lett., 1977, 18, 837 CrossRef.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr., G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 992086. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03000a |
| ‡ Present address: Department of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. |
| This journal is © The Royal Society of Chemistry 2014 |