 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent, sequence-specific attachment of long DNA molecules to a surface using DNA-templated click chemistry†
Gary R.
Abel
Jr.
,
Blessing Huynh
Cao
,
Jason E.
Hein
and
Tao
Ye
*
Chemistry and Chemical Biology, School of Natural Sciences, University of California, 5200 N. Lake Rd., Merced, CA 95343, USA. E-mail: tao.ye@ucmerced.edu
First published on 6th June 2014
Abstract
We present a novel method that covalently and sequence-specifically attaches long DNA molecules to a surface that is compatible with high-resolution atomic force microscopy (AFM) imaging. Surfaces prepared with this approach are ideally suited for performing biophysical experiments on single DNA molecules.
Techniques that probe DNA at the single molecule level have provided unique insight into fundamental biological processes, such as gene expression and DNA–protein binding. For many techniques, including AFM force spectroscopy,1 optical tweezers,2 and single molecule fluorescence,3 it is necessary for a long DNA molecule (>100 bp) to be attached at one end to a solid support. The prevalence of surface-tethering can be attributed to the following: tethering restricts the diffusion of the molecule, allowing for extended observation; it isolates the molecule, preventing unwanted interactions with neighboring molecules; and it provides a convenient handle with which the molecule can be manipulated. Going beyond existing approaches, the capability of attaching long DNA molecules with different sequences to specific locations on the surface would potentially allow researchers to perform high throughput, multiplexed single molecule measurements on spatially addressable nanoarrays.4 Among the existing strategies to tether DNA to a solid support, such as biotin–avidin recognition,2,3 amide coupling,1 and gold–thiol chemistry,5 several limitations remain. Although covalent attachment methods are effective at tethering relatively short DNA to a solid support, they become significantly slower for longer DNA due to orientational constraints and diffusion limitation of the reaction.6 Existing covalent attachment chemistries are also not sequence specific, i.e., cannot selectively tether molecules with different sequences, which is important for nanoarrays. Often the surface has significant heterogeneity in topography and chemical functionality, resulting in nonspecific interactions that introduce artefacts into measurements. Very little is known about the heterogeneity in the local environment of the immobilized molecules, as the surfaces are generally not compatible with high-resolution AFM imaging, the only technique that is potentially capable of characterizing these surfaces with sub 10 nm resolution.7,8 Typically, the surface interactions are either too strong to allow for the conformational freedom that is needed for unperturbed reactions, or too weak to immobilize the molecules for imaging.7,8 Existing AFM studies have relied on exchanging the buffer solution to switch the binding affinities between DNA and the mica substrate to allow for conformational freedom in a weakly bound state and imaging in a strongly bound state.9,10 However, the biochemical reactions are heavily perturbed by surface interactions even in the weakly bound state, as there must be sufficient surface interactions to prevent irreversible desorption of DNA.10
Here we demonstrate that long DNA can be covalently and sequence-specifically tethered to a surface that is compatible with both high-resolution single molecule imaging and unimpeded biomolecular interactions. This is accomplished by attaching short DNA ‘anchor’ strands that bear an alkyne group to a surface that can switch DNA–surface interactions on demand, allowing the anchor strands to capture long ‘target’ DNA with a complementary sequence, and then covalently crosslinking the strands via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) (Fig. 1). Base-pairing interactions have been previously used to bring reactive groups into close proximity to facilitate reactions in a homogeneous solution phase.13,14 Here we show that the specificity and greater efficiency of the base-pairing process can guide the covalent attachment of long DNA strands to our switchable surface. This novel approach may have broad utility in single molecule techniques that require surface-tethering of long DNA, and may enable the production of single molecule nanoarrays.
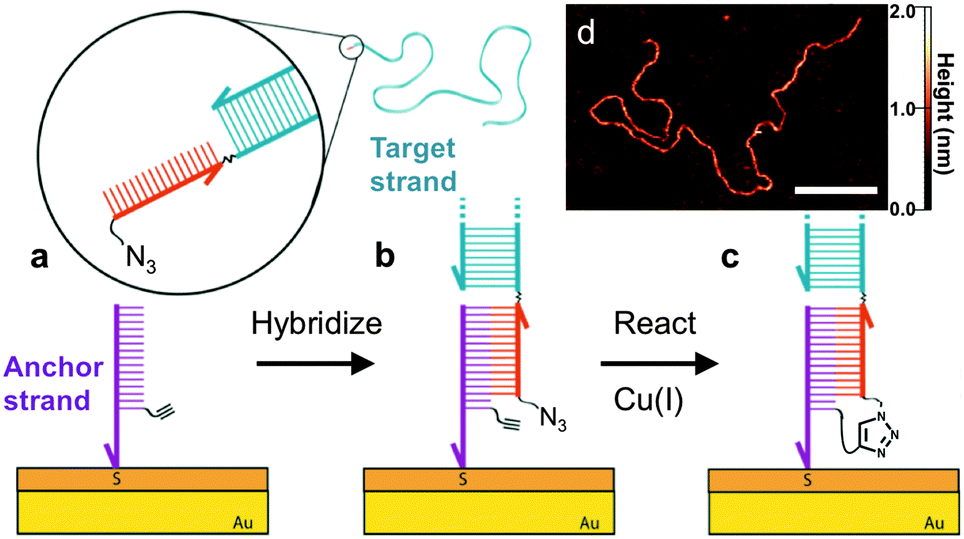 | ||
| Fig. 1 Schematic illustration of the DNA-templated surface coupling reaction. (a) The thiolated anchor strands bearing alkyne groups (purple) are inserted into the defects of a self-assembled monolayer on a gold substrate.7,11,12 The surface is then exposed to the target strand (blue) containing a complementary single-stranded tail (red) with a terminal azide group. (b) Upon hybridization of the target and anchor strands, the azide and alkyne groups are placed in close proximity. (c) Cu(I) catalyzes the crosslinking reaction, leaving the target DNA covalently tethered to the surface. (d) AFM image of a 3679 base-pair (bp) DNA strand that has been attached using this approach. Scale bar is 100 nm. | ||
We employed the CuAAC reaction to crosslink the DNA strands because it is efficient and orthogonal to a wide range of biochemical reactions.15 Prior to performing the reaction on the surface, we tested the approach by crosslinking two short, complementary DNA strands in solution. This step was useful not only for identifying conditions that would provide a suitable reaction yield, but also for assessing the extent of damage to the DNA by the presence of copper. It has been shown that the in situ generation of Cu(I) catalyst by ascorbate reduction of Cu(II) can damage DNA by generating reactive oxygen species.16 Protective Cu-binding ligands have been used in bioconjugation reactions to minimize the oxidative damage,17 and it was found previously that the structure of the ligand can profoundly affect both the reactivity and biocompatibility of the Cu catalyst.18 We compared the performance of TBTA, a ligand commonly used in the literature, with two alternative ligands that show improved aqueous solubility and an increased reaction rate (Fig. 2). To perform the coupling reaction in solution, the two strands were first hybridized by heating to 70 °C and slowly cooling to room temperature in a hybridization buffer. Next the Cu(I) solution was added to initiate the reaction, and aliquots of the solution were removed at predetermined time points and added to a quenching buffer to stop the reaction. The yield at different time points was assessed by denaturing polyacrylamide gel electrophoresis (D-PAGE). As shown in Fig. 2, the reactants appeared as two separate bands due to different mobilities in the gel. The product, corresponding to the crosslinked DNA, appeared as a third, slower moving band, and was easily distinguished from the reactant bands. By using the normalized intensity of the product band, we were able to quantify the yield of the crosslinking reaction. D-PAGE was also useful for monitoring the extent of damage due to reactive oxygen species, which led to streaking in the gel and a decrease in the product band intensity (ESI†). By varying conditions such as Cu(II) and ascorbate concentrations, solvent, ligand type, and ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu ratio, and measuring the resulting reaction yield, we identified a set of conditions in which the reaction was apparently complete within 5 minutes with minimal degradation of the DNA.
:Cu ratio, and measuring the resulting reaction yield, we identified a set of conditions in which the reaction was apparently complete within 5 minutes with minimal degradation of the DNA.
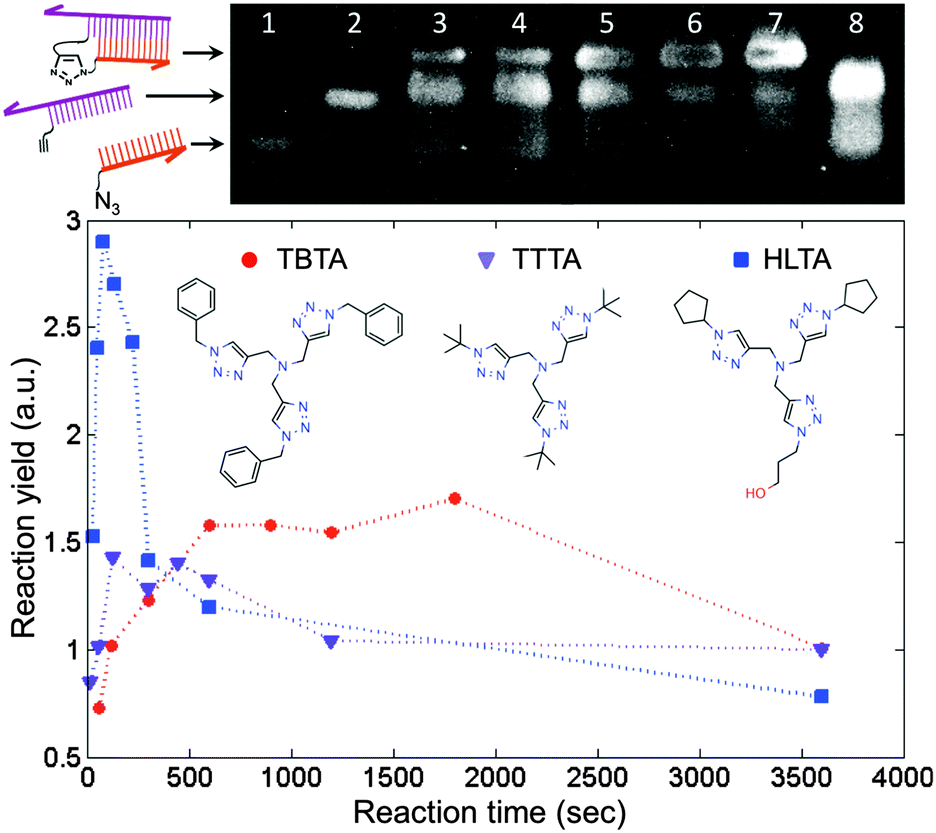 | ||
| Fig. 2 Use of denaturing polyacrylamide gel electrophoresis to quantify the DNA-templated crosslinking reaction yield in solution. (top) Sample gel image. Lanes 1 and 2 are the target and anchor strands, respectively. Lanes 3–7 are a series of time points from a mixture of both strands after hybridization and reaction with the Cu(I) catalyst under one set of conditions tested. Lane 8 is a control where the Cu(I) catalyst was omitted. (bottom) Reaction yield with three copper-chelating ligands as a function of time. The decrease in yield at longer times can be attributed to oxidative degradation of the DNA. We found that degradation could be significantly reduced by minimizing the amount of oxygen present and by using an excess of the ligand. | ||
After determining suitable reaction conditions in solution, we tested whether the coupling reaction could be used to covalently and sequence-specifically crosslink long DNA to the surface. As demonstrated in Fig. 3, our method consists of a simple three-step approach in which the long target DNA strand binds to, and then reacts with, a short, complementary anchor oligonucleotide on the surface. First the thiolated anchor strands are inserted into surface defects of a self-assembled monolayer (SAM) on a gold substrate.8 Next the anchor surface is exposed to a solution containing the target DNA strands, which then hybridize with the surface anchors. Importantly, only strands bearing a single-stranded tail with the complementary sequence will bind during this step. Once hybridization has occurred, the surface is rinsed and exposed to a catalytic Cu(I) solution. At this point the target strands should be covalently crosslinked to the surface by the triazole product.
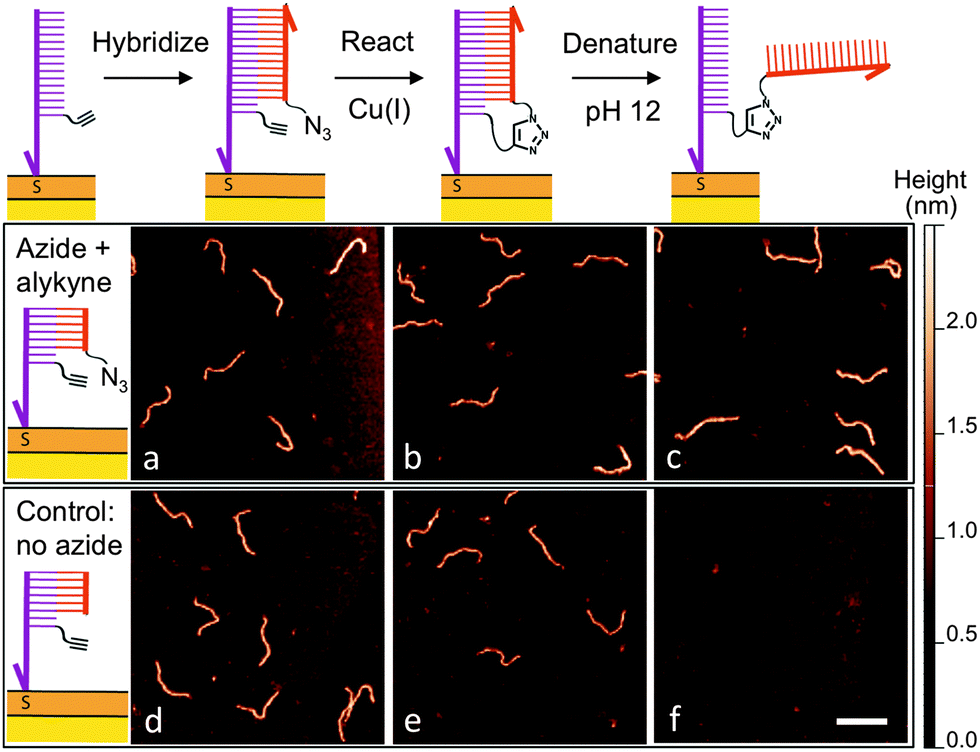 | ||
| Fig. 3 AFM images taken after each step of attaching 396 bp DNA to the surface. (a) The azide-labeled DNA targets are hybridized with surface anchor strands containing an alkyne group. (b) Next the surface is exposed to Cu(I), catalyzing the formation of the triazole product. (c) To verify that the DNA is covalently crosslinked, the surface is exposed to alkaline conditions so that any unreacted DNA is denatured and rinsed away. (d–f) The same hybridization, reaction, and denaturation steps, respectively, but with the azide group omitted. In this case, >99% of the DNA was removed after denaturing. Scale bar is 100 nm. | ||
High-resolution AFM imaging would be a powerful means to directly measure the conformation and spatial distribution of individual biomolecules attached to surfaces, as well as the molecular scale morphology of their local environments. However, high-resolution imaging has not been possible on most existing biofunctional surfaces due to the weak surface interaction that is required to allow unimpeded biomolecular reactions and prevent nonspecific adsorption. Here, we overcome this challenge by covalently attaching the DNA to a carboxyl-terminated SAM.8 The covalent anchoring and precise SAM surface chemistry allow us to use electrostatic repulsion to lift up the anchored DNA into a solution-like environment. In contrast to existing studies on mica, we no longer have to compromise between minimizing the surface interaction to allow reactions and ensuring sufficient binding to prevent irreversible desorption.9 Imaging was then performed in a divalent cation buffer that strongly immobilizes the DNA due to charge inversion of the surface.8 As shown in Fig. 3, after hybridizing with the surface anchor strands, the 396 bp DNA target strands appear as 2 nm tall worm-like features with the expected contour length of ∼120 nm; hence hybridization is facile on this surface. The surface was then exposed to a catalyst solution containing Cu(II), the sodium ascorbate reducing agent, and a Cu(I)-binding ligand. The kinetics of the surface reaction is slower than that in a homogeneous solution and an increased reaction time of 25 minutes was used to ensure a good yield. AFM imaging revealed that the DNA was still present on the surface after the reaction. Statistical analysis of the DNA chain conformations before and after the reaction indicates that oxidative degradation of the DNA during this step is minimal (Fig. S7, ESI†). To verify covalent crosslinking, the surface was next exposed to an alkaline NaOH solution (pH 12), which is known to disrupt the hydrogen bonds in base pairs and denature double-stranded DNA.19 The majority of the molecules remained bound to the surface, consistent with the DNA being linked to the surface through a covalent bond in addition to base pairing. By counting molecules over a large area before and after denaturation, we determined the yield of the crosslinking reaction to be 83%. Note that the denaturing pH was selected such that the base pairing between the single-stranded tail and the anchor strand is melted (Fig. 3c) but the double-stranded segment of the target DNA remains intact due to its higher melting temperature. When a higher pH was used, the anchored target strands were completely melted into single-stranded DNA (Fig. S12, ESI†). In control experiments where either the azide or alkyne group is excluded, the targets indeed hybridized with the surface anchors, and were still present on the surface after exposure to the Cu(I) catalyst. However, after exposure to denaturing conditions, >99% of the DNA molecules were rinsed away, confirming that base-pairing alone is not sufficient for stable attachment to the surface under these conditions (Fig. 3f and Fig. S10, ESI†). Finally, to verify the sequence-specificity, we repeated the experiment using sequence-mismatched DNA anchors and target strands containing the alkyne and azide groups. None of the mismatched DNA was found on the surface after hybridization, confirming that the crosslinking reaction can be programmed by the base sequence (Fig. S11, ESI†). Using the above strategy, we have attached both single- and double-stranded DNA ranging in size from 396 to 3679 bases to the surface.
Our new approach to link long DNA molecules to highly ordered SAMs will enable AFM studies of biochemical reactions that are difficult on traditional mica substrates. In addition, it potentially has broad utility in other single molecule techniques2,3 because it can reduce measurement artefacts that are caused by nonspecific surface interactions and heterogeneity in the local environment. Moreover, our method allows long DNA that are difficult to pattern directly to be crosslinked to short anchor strands that are readily patterned with existing techniques.20–22 Therefore, it will pave the way for spatially addressable nanoarrays that can allow the parallel measurement of a large number of single molecules.
The authors acknowledge financial support from the National Science Foundation (CHE-1048651 and CHE-1361066) and UC Cancer Research Coordinating Committee.
Notes and references
- M. Erdmann, R. David, A. Fornof and H. E. Gaub, Nat. Nanotechnol., 2010, 5, 154 CrossRef CAS PubMed.
- K. M. Herbert, W. J. Greenleaf and S. M. Block, Annu. Rev. Biochem., 2008, 77, 149 CrossRef CAS PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Acc. Chem. Res., 2005, 38, 542 CrossRef CAS PubMed.
- S. Howorka and J. Hesse, Soft Matter, 2014, 10, 931 RSC.
- G. J. L. Wuite, S. B. Smith, M. Young, D. Keller and C. Bustamante, Nature, 2000, 404, 103 CrossRef CAS PubMed.
- S. C. Huang, M. D. Stump, R. Weiss and K. D. Caldwell, Anal. Biochem., 1996, 237, 115 CrossRef CAS PubMed.
- E. A. Josephs and T. Ye, J. Am. Chem. Soc., 2012, 134, 10021 CrossRef CAS PubMed.
- G. R. Abel Jr, E. A. Josephs, N. Luong and T. Ye, J. Am. Chem. Soc., 2013, 135, 6399 CrossRef PubMed.
- N. Thomson, S. Kasas, B. Smith, H. Hansma and P. Hansma, Langmuir, 1996, 12, 5905 CrossRef.
- M. Guthold, X. Zhu, C. Rivetti, G. Yang, N. H. Thomson, S. Kasas, H. G. Hansma, B. Smith, P. K. Hansma and C. Bustamante, Biophys. J., 1999, 77, 2284 CrossRef CAS.
- E. A. Josephs and T. Ye, Nano Lett., 2012, 12, 5255 CrossRef CAS PubMed.
- E. A. Josephs and T. Ye, ACS Nano, 2013, 7, 3653 CrossRef CAS PubMed.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848 CrossRef CAS PubMed.
- P. Kocalka, A. H. El-Sagheer and T. Brown, ChemBioChem, 2008, 9, 1280 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- P. Y. Liu, N. Jiang, J. Zhang, X. Wei, H. H. Lin and X. Q. Yu, Chem. Biodiversity, 2006, 3, 958 CAS.
- Q. Wang, T. R. Chan, R. Hilgraf, V. R. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570 CrossRef CAS PubMed.
- M. Ageno, E. Dore and C. Frontali, Biophys. J., 2009, 9, 1281 CrossRef.
- L. M. Demers, D. S. Ginger, S. J. Park, Z. Li, S. W. Chung and C. A. Mirkin, Science, 2002, 296, 1836 CrossRef CAS PubMed.
- S. K. Kufer, E. M. Puchner, H. Gumpp, T. Liedl and H. E. Gaub, Science, 2008, 319, 594 CrossRef CAS PubMed.
- E. A. Josephs and T. Ye, J. Am. Chem. Soc., 2010, 132, 10236 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, mass spectrometry and NMR data, and additional AFM results. See DOI: 10.1039/c4cc02900c |
| This journal is © The Royal Society of Chemistry 2014 |