 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designed cell penetrating peptide dendrimers efficiently internalize cargo into cells†
Gabriela A.
Eggimann‡
,
Emilyne
Blattes‡
,
Stefanie
Buschor
,
Rasomoy
Biswas
,
Stephan M.
Kammer
,
Tamis
Darbre
* and
Jean-Louis
Reymond
*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch; Fax: +41 31 631 80 57; Tel: +41 31 631 43 25
First published on 15th May 2014
Abstract
Redesigning linear cell penetrating peptides (CPPs) into a multi-branched topology with short dipeptide branches gave cell penetrating peptide dendrimers (CPPDs) with higher cell penetration, lower toxicity and hemolysis and higher serum stability than linear CPPs. Their use is demonstrated by delivering a cytotoxic peptide and paclitaxel into cells.
The cell membrane poses an impermeable barrier for a large number of compounds, in particular peptides, limiting their use as drugs. Nevertheless the discovery of the cell penetrating properties of the HIV-1 Tat protein1 led to the discovery of a variety of cell penetrating peptides (CPPs),2 an effect which was later also reported for synthetic oligomer analogs of linear peptides,3 cyclic peptides,4 and various organic dendrimers.5 CPPs can serve as carriers for drug delivery, however they suffer from the typical metabolic instability of linear peptides and tend to be hemolytic and cytotoxic. Interestingly the cell internalization of CPPs can be enhanced by grafting them onto multivalent scaffolds, but their toxicity is usually also increased in such constructs despite the fact that their folding propensity is retained.6 We showed recently that peptide dendrimers containing very short mono- or dipeptide branches,7 while not able to form stable secondary structures,7b are generally more resistant to proteolytic degradation compared to linear peptides,7c and show almost no cytotoxicity when used for DNA transfection7f and only very weak hemolysis when designed as antimicrobials,7e which is probably a benefit of their particular molten globule conformation enforced by the dendritic topology.7a Although no increase in cellular uptake was reported with branched octaarginines compared to linear (Arg)8,8 we asked the question whether a broader survey of CPPs redesigned in a multi-branched topology might lead to cell penetrating peptide dendrimers (CPPDs) with higher cellular uptake, metabolic stability and lower toxicity compared to linear CPPs, and potential use as drug delivery agents.
Fourteen peptide dendrimers were prepared by SPPS with sequence (X8X7)8(KX6X5)4(KX4X3)2KX2X1K* (K = branching lysine) and labeled with 5(6)-carboxyfluorescein (CF) attached to the ε-amino group of the core lysine residue (K*) (Scheme 1). Variable residues (X) were assigned to amino acids found in the linear cationic CPP Tat1 or the amphipathic CPPs TP109 and pVEC10 to form dendrimers with Arg and Lys (D1–D4 and D11) or only Lys (D5–D10) as cationic residues, featuring various ratios and distribution of cationic and hydrophobic side chains in the branches and optional acetylation of N-termini to reduce the number of positive charges (Table 1).
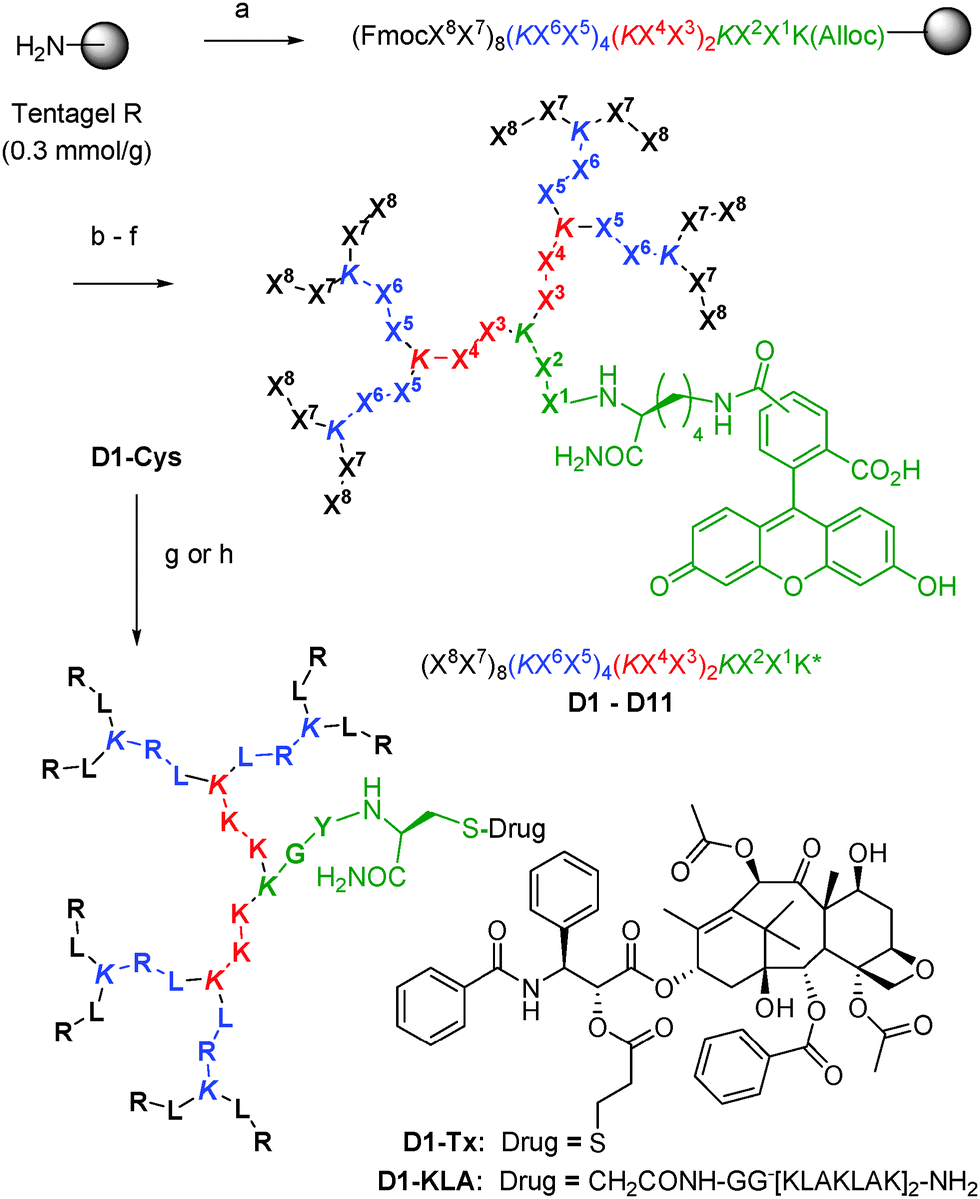 | ||
| Scheme 1 Synthesis of CPPD. Conditions: (a) Fmoc solid-phase peptide synthesis (SPPS), Xi = amino acid, K = branching lysine; (b) PhSiH3, Pd(PPh3)4; (c) 5(6)-carboxyfluorescein, HOBt, DIC; (d) piperidine/DMF; (e) optional: Ac2O; (f) CF3CO2H/iPr3SiH/H2O, then preparative HPLC; (g) ClCH2CO-GG[KLAKLAK]2NH2, DIEA; (h) PTX-PDP, DIEA. See ESI† for details. | ||
| Cpd. | Sequencea | Yieldb (mg (%)) | +c | Arg | Lys | Hyd.d | MFI av. (1 μM) | (10 μM)e | Hemolysisf (μg mL−1/μM) |
|---|---|---|---|---|---|---|---|---|---|
| HeLa, CHO, Jurkat | |||||||||
| a One letter codes for amino acids. Branching diamino acids are shown in italics. * = 5(6)-carboxyfluorescein amidated to the Lys side chain (K*) or the N-terminus (*-) added as the last coupling step, see ESI for details. Ac = acetyl. All peptides are carboxamide (CONH2) at the C-terminus. b Yields given for RP-HPLC purified products as TFA salts. c + = number of cationic amino acid side chains (Lys and Arg) and free N-termini. d Hyd = hydrophobic residues: Ala, Ile, Leu and Tyr. e Flow cytometry data. MFI average = average of the mean fluorescence intensity of the tested peptides over background. Three independent experiments are used to get values. f Hemolysis assay detailed in ESI. n.d. = not determined. | |||||||||
| Tat | *-YGRKKRRQRRR | 90.9 (26) | 8 | 6 | 2 | 1 | 2, 2, 11 | 14, 23, 65 | 125/44 |
| D1 | (RL)8(KRL)4(KKK)2KGYK* | 14.7 (3) | 24 | 12 | 4 | 13 | 11, 10, 49 | 214, 114, 1450 | 31/4 |
| D2 | (RL)8(KLL)4(KKK)2KGYK* | 9.6 (6) | 20 | 8 | 4 | 17 | 20, 22, 108 | 158, 175, 631 | 31/4 |
| D3 | (RL)8(KNN)4(KKK)2KGYK* | 23.3 (5) | 20 | 8 | 4 | 9 | 5, 4, 40 | 64, 48, 210 | 125/17 |
| D4 | (GY)8(KKR)4(KQQ)2KRRK* | 13.2 (2) | 18 | 6 | 4 | 12 | 3, 4, 27 | 19, 51, 112 | 63/9 |
| AcD4 | (AcGY)8(KKR)4(KQQ)2KRRK* | 4.7 (1) | 10 | 6 | 4 | 12 | 3, 2, 9 | 15, 25, 65 | 250/38 |
| TP10 | *-AGYLLGKINLKALAALAKKIL | 51.3 (15) | 4 | — | 4 | 15 | 7, 6, 23 | 152, 186, 385 | 31/10 |
| D5 | (AL)8(KIK)4(KLA)2KKIK* | 33.4 (5) | 13 | — | 5 | 25 | 4, 9, 28 | 46, 122, 118 | 63/11 |
| D6 | (LA)8(KKL)4(KKL)2KYAK* | 55.1 (8) | 14 | — | 6 | 24 | 2, 5, 7 | 17, 21, 31 | 1000/163 |
| AcD6 | (AcLA)8(KKL)4(KKL)2KYAK* | 9.0 (7) | 6 | — | 6 | 24 | 2, 7, 14 | 35, 40, 64 | n.d. |
| D7 | (IK)8(KLA)4(KLA)2KKIK* | 6.4 (1) | 17 | — | 9 | 21 | 5, 14, 29 | 33, 51, 355 | 125/19 |
| AcD7 | (AcIK)8(KLA)4(KLA)2KKIK* | 3.0 (1) | 9 | — | 9 | 21 | 5, 14, 190 | 33, 259, 862 | 63/10 |
| D8 | (LA)8(KIK)4(KLA)2KKIK* | 11.1 (2) | 13 | — | 5 | 25 | 2, 4, 11 | 20, 30, 71 | 63/11 |
| D9 | (LA)8(KLA)4(KIK)2KKIK* | 8.2 (2) | 11 | — | 3 | 27 | 2, 2, 10 | 14, 14, 61 | 125/22 |
| D10 | (LA)8(KLA)4(KLA)2KKIK* | 12.3 (3) | 9 | — | 1 | 29 | 3, 2, 5 | 9, 8, 36 | 250/48 |
| pVEC | *-LLIILRRRIRKQAHAHSK | 94.3 (26) | 6 | 4 | 2 | 8 | 36, 71, 84 | 126, 210, 292 | 4/1 |
| D11 | (LI)8(KRK)4(KRA)2KHSK* | 15.0 (2) | 18 | 6 | 4 | 18 | 3, 12, 58 | 60, 106, 968 | 500/70 |
| AcD11 | (AcLI)8(KRK)4(KRA)2KHSK* | 7.0 (5) | 10 | 6 | 4 | 18 | 4, 8, 26 | 44, 105, 385 | n.d. |
CD spectra of CPPDs show random coil signals independent of amino acid composition consistent with the disordered conformation of branched peptides and similar to Tat and pVEC (Fig. 1a and Fig. S1, ESI†). CPPD uptake by HeLa, CHO and Jurkat cells was evaluated by flow cytometry after 1 h incubation of the cells with 1 μM or 10 μM compounds (Table 1, Fig. 1b–d and Fig. S2, ESI†). A strong fluorescence increase was observed with arginine containing dendrimers D1 and D2 (staining 10–15 times higher than the parent linear Tat), D11 and AcD11 (staining comparable to the parent linear pVEC). Exchanging the eight leucines in the G2-branches of D2 for polar asparagines to form D3 decreased the MFI average in spite of the same total number of cationic residues (8 Arg and 4 Lys), highlighting the importance of hydrophobic residues for uptake. D5 and AcD7 with only lysine as a cationic residue also showed strong fluorescence comparable to the parent linear TP10, implying that arginine is not necessary for cellular uptake of CPPD. The remaining CPPDs showed MFI average comparable to linear Tat. Hydrophobic D10 with only one cationic side chain was the least efficient CPPD in the library.
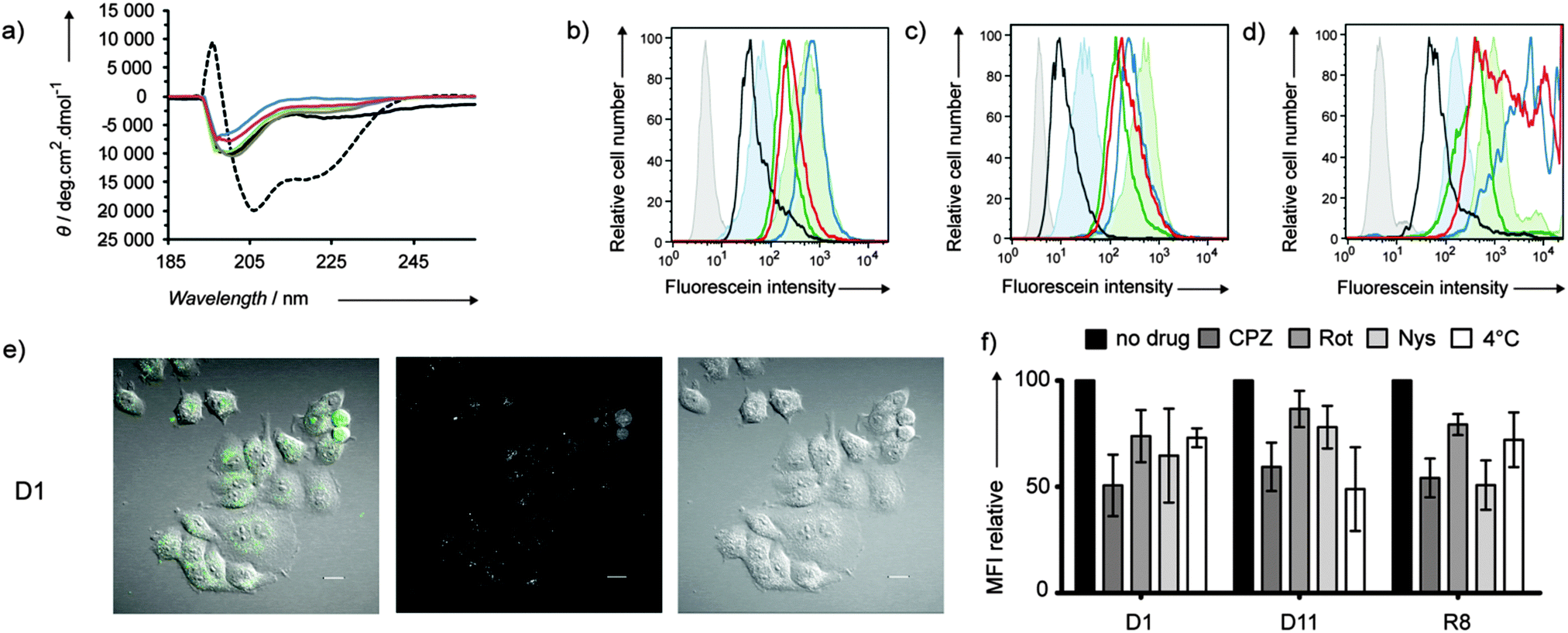 | ||
| Fig. 1 (a) CD measurements of D1 (blue), D5 (green), D10 (grey), and D11 (red). Tat (black), TP10 (dotted black) in PBS buffer (pH 7.4). (b–d) Flow cytometry histograms for HeLa (b), CHO (c) and Jurkat (d) cells after 1 h incubation with 10 μM of peptide dendrimers at 37 °C. D1 (blue), D5 (green), D10 (black), and D11 (red). Untreated cells (grey), Tat (filled blue), TP10 (filled green). (e) Observed uptake of D1 (10 μM) after 1 h incubation at 37 °C on HeLa cells in live confocal microscopy of the differential interference contrast (DIC, right panel) and the CF fluorescence at 525 nm (middle panel). The left panel shows the merged images of the DIC and fluorescence pictures. White bar = 20 μm. See Fig. S5 (ESI†) for data with CPPD D11. (f) Uptake levels of D1 and D11 in HeLa cells in the presence of various inhibitors. Cells were pretreated 30 min with 50 μM chlorpromazine (CPZ), 20 μM rottlerin (Rot), 25 μg mL−1 nystatin (Nys) or cooled down at 4 °C prior to 1 h incubation with 10 μM CPPD or linear octaarginine (R8) in the presence of an inhibitor. Error bars represent the SD of three independent experiments. See also Fig. S6 (ESI†) for CHO cells. | ||
Fixed confocal microscopy images with HeLa and CHO cells confirmed the trends seen in flow cytometry and revealed the cellular localization of peptide dendrimers (Fig. S3, ESI†). For the Arg containing dendrimers D1, D2, D3 and D11, and D5 with only Lys as a cationic residue, images showed a strong staining of the cytoplasm similar to Tat in both cell lines. In contrast to PAMAM dendrimers,5b acetylation of N-terminal amino groups did not block cellular uptake of CPPDs, although differences of subcellular localization were visible between D11 (homogeneous staining) and AcD11 (punctuated patterns). Live confocal microscopy experiments were performed with D1 and D11 to confirm cellular uptake and distribution (Fig. 1e, Fig. S5 and S6, ESI†). After 1 h, dendrimers were found inside cells localized in the cytoplasm in a punctuated pattern suggesting confinement in endosomes.
Both energy-dependent endocytosis and energy-independent or direct translocation across the cell membrane have been proposed as uptake mechanisms for linear CPPs.11 The uptake of dendrimer D1 by HeLa cells was reduced by chlorpromazine, indicating clathrin dependent uptake, and to a lesser extent by rottlerin (macropinocytosis inhibitor) and nystatin (caveola/lipid raft-dependent endocytosis inhibitor) (Fig. 1f). Uptake was only 30% lower at 4 °C compared to that at 37 °C, suggesting that direct membrane translocation also takes place for D1. Dendrimer D11 showed a similar pattern however with reduced uptake at 4 °C, indicating lower self-translocating ability probably due to its smaller number of positive charges and different distribution of Arg residues in the branches compared to D1. Results with CHO cells (Fig. S6, ESI†) show primarily a clathrin dependent uptake for both D1 and D11 and a reduced uptake at 4 °C (energy-dependent uptake mostly) in line with previous observations that the cell type plays a critical role in the internalization mechanism of CPPs. In the case of CPPDs, clathrin mediated uptake appears to be the major process of internalization taking place in both cell lines.
CPPDs were not significantly toxic to HeLa or CHO cells (up to 10 μM, 24 h, Fig. S4, ESI†). However less than 50% viable Jurkat cells remained after 24 h exposure to 10 μM pVEC, D1, D2, D7, D11 or AcD11, which belong to the most cell penetrating compounds. While hemolysis was very strong for the linear CPP pVEC (4 μg mL−1), CPPDs were less hemolytic including those with strong cell penetrating properties (30–500 μg mL−1, Table 1).
Dendrimers D1 and D11 combined efficient uptake into cells with moderate toxicity and were further investigated. Degradation experiments in human serum showed that 40% of D1 and D11 were still unchanged after 12 h while linear Tat was completely degraded, in line with previous reports that peptide dendrimers are more resistant to proteolysis than linear peptides (Fig. S7, ESI†).7d–f To evaluate the ability of D1 and D11 to deliver cargos into cells, dendrimers were conjugated via a thioether bridge to the α-helical tetradecapeptide [KLAKLAK]2 (KLA), an antimicrobial peptide that does not penetrate mammalian cells but induces cell death by disrupting the mitochondrial membrane if delivered into the cytosol.12 The resulting conjugates D1-KLA and D11-KLA (Scheme 1) were found to be significantly more cytotoxic to HeLa and CHO cells compared to Tat-KLA at 10 μM (Fig. S8 and S9, ESI†), while KLA alone or an ungrafted dendrimer were not toxic (Table 2, Fig. S10, ESI†). Covalent conjugation to CPPD does not change the bioactivity of KLA since disulfide bridged conjugates to both D1 and D11 showed cytotoxicity similar to the thioether adducts (Fig. S11, Table S1, ESI†).
| Cpd. | IC50 HeLa cells | IC50 CHO cells |
|---|---|---|
| a Cell survival measured after 24 h (KLA series) or 72 h (paclitaxel series) with the WST-8 assay. b % of surviving cells at the indicated concentration, which was the highest measured. No IC50 is given when the full inhibition curve could not be measured. c With 1.6% DMSO added to solubilise paclitaxel. | ||
| KLA | ≫20 μM (100%)b | ≫20 μM (93%)b |
| D1-KLA | 4.6 ± 0.2 μM | 7.8 ± 0.6 μM |
| D11-KLA | 6.0 ± 0.3 μM | 8.9 ± 0.6 μM |
| Tat-KLA | 10.1 ± 0.5 μM | ∼20 μM (48%)b |
| Paclitaxel (PTX) | 8.4 ± 0.6 nMc | ∼4 μM (60%)b |
| D1-PTX | 47 ± 15 nM | ∼4 μM (57%)b |
| D11-PTX | 41 ± 9 nM | ∼4 μM (46%)b |
In a further example, D1 and D11 were conjugated to paclitaxel (PTX), a notoriously insoluble anticancer drug whose solubility and targeting can be significantly improved by incorporation into a variety of nanocarriers including dendrimers and peptides.13 CPPD conjugates D1-PTX and D11-PTX were prepared by disulfide bond formation between D1-Cys or D11-Cys and paclitaxel-2′-(3-(2-pyridyldithio)) propionate (PTX-PDP, Scheme 1). The dithiopropionyl linker has been reported for PTX-octaarginine14 and for dendrimer drug conjugates15 and is susceptible to intracellular reductive cleavage. Our CPPD-PTX conjugates were water soluble and stable in cell culture over the time of the experiment, yet retained most of the selective cytotoxicity of PTX to HeLa cells over CHO cells (Table 2, Fig. S12, ESI†).
In summary, redesigning CPPs into G3 peptide dendrimers with short dipeptide branches gave CPPDs with stronger cellular uptake, lower cytotoxicity and hemolysis, and higher stability towards serum degradation compared to their linear counterpart. Cellular uptake was observed in a diversity of sequences containing either Arg or Lys as a cationic residue and a balanced ratio of hydrophobic residues. Dendrimer D1 inspired by the Tat peptide and D11 inspired by pVEC efficiently localized in the cytoplasm and delivered cytotoxic cargo into cells. The low intrinsic toxicity and hemolysis of CPPDs might result from their inability to fold into amphipathic α-helical membrane lytic aggregates. The higher cellular uptake of CPPDs compared to linear CPPs probably reflects in part their larger size, which is remarkably obtained without increased synthetic complexity since the described CPPDs are obtained in only 12 SPPS coupling steps warranting low production costs. Due to their ease of synthesis and favorable properties CPPDs represent a promising and versatile new class of cell penetrating devices.
This work was supported financially by the University of Berne and the Swiss National Science Foundation.
Notes and references
- (a) S. Fawell, J. Seery, Y. Daikh, C. Moore, L. L. Chen, B. Pepinsky and J. Barsoum, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 664–668 CrossRef CAS; (b) E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef PubMed.
- (a) S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed; (b) S. B. Fonseca, M. P. Pereira and S. O. Kelley, Adv. Drug Delivery Rev., 2009, 61, 953–964 CrossRef CAS PubMed; (c) E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- (a) I. Peretto, R. M. Sanchez-Martin, X. H. Wang, J. Ellard, S. Mittoo and M. Bradley, Chem. Commun., 2003, 2312–2313 RSC; (b) J. H. Moon, W. McDaniel, P. Maclean and L. F. Hancock, Angew. Chem., Int. Ed., 2007, 46, 8223–8225 CrossRef CAS PubMed; (c) E. R. Gillies, F. Deiss, C. Staedel, J. M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081–4084 CrossRef CAS PubMed; (d) C. B. Cooley, B. M. Trantow, F. Nederberg, M. K. Kiesewetter, J. L. Hedrick, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2009, 131, 16401–16403 CrossRef CAS PubMed; (e) E. I. Geihe, C. B. Cooley, J. R. Simon, M. K. Kiesewetter, J. A. Edward, R. P. Hickerson, R. L. Kaspar, J. L. Hedrick, R. M. Waymouth and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13171–13176 CrossRef CAS PubMed; (f) U. Sternberg, E. Birtalan, I. Jakovkin, B. Luy, U. Schepers, S. Brase and C. Muhle-Goll, Org. Biomol. Chem., 2013, 11, 640–647 RSC.
- (a) Y. U. Kwon and T. Kodadek, Chem. Biol., 2007, 14, 671–677 CrossRef CAS PubMed; (b) G. Lattig-Tunnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed; (c) D. Mandal, A. Nasrolahi Shirazi and K. Parang, Angew. Chem., Int. Ed., 2011, 50, 9633–9637 CrossRef CAS PubMed; (d) Z. Qian, T. Liu, Y. Y. Liu, R. Briesewitz, A. M. Barrios, S. M. Jhiang and D. Pei, ACS Chem. Biol., 2013, 8, 423–431 CrossRef CAS PubMed.
- (a) B. Aussedat, E. Dupont, S. Sagan, A. Joliot, S. Lavielle, G. Chassaing and F. Burlina, Chem. Commun., 2008, 1398–1400 RSC; (b) L. Albertazzi, M. Serresi, A. Albanese and F. Beltram, Mol. Pharmaceutics, 2010, 7, 680–688 CrossRef CAS PubMed; (c) R. J. Amir, L. Albertazzi, J. Willis, A. Khan, T. Kang and C. J. Hawker, Angew. Chem., Int. Ed., 2011, 50, 3425–3429 CrossRef CAS PubMed.
- (a) K. Sheldon, D. Liu, J. Ferguson and J. Gariepy, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2056–2060 CrossRef CAS; (b) K. S. Kawamura, M. Sung, E. Bolewska-Pedyczak and J. Gariepy, Biochemistry, 2006, 45, 1116–1127 CrossRef CAS PubMed; (c) A. M. Angeles-Boza, A. Erazo-Oliveras, Y.-J. Lee and J.-P. Pellois, Bioconjugate Chem., 2010, 21, 2164–2167 CrossRef CAS PubMed; (d) G. A. Eggimann, S. Buschor, T. Darbre and J.-L. Reymond, Org. Biomol. Chem., 2013, 11, 6717–6733 RSC.
- (a) S. Javor, E. Delort, T. Darbre and J. L. Reymond, J. Am. Chem. Soc., 2007, 129, 13238–13246 CrossRef CAS PubMed; (b) S. Javor and J. L. Reymond, J. Org. Chem., 2009, 74, 3665–3674 CrossRef CAS PubMed; (c) P. Sommer, V. S. Fluxa, T. Darbre and J. L. Reymond, ChemBioChem, 2009, 10, 1527–1536 CrossRef CAS PubMed; (d) J. L. Reymond and T. Darbre, Org. Biomol. Chem., 2012, 10, 1483–1492 RSC; (e) M. Stach, N. Maillard, R. U. Kadam, D. Kalbermatter, M. Meury, M. G. P. Page, D. Fotiadis, T. Darbre and J.-L. Reymond, MedChemComm, 2012, 3, 86–89 RSC; (f) A. Kwok, G. A. Eggimann, J. L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- S. Futaki, I. Nakase, T. Suzuki, Z. Youjun and Y. Sugiura, Biochemistry, 2002, 41, 7925–7930 CrossRef CAS PubMed.
- (a) M. Pooga, M. Hallbrink, M. Zorko and U. Langel, FASEB J., 1998, 12, 67–77 CAS; (b) U. Soomets, M. Lindgren, X. Gallet, M. Hällbrink, A. Elmquist, L. Balaspiri, M. Zorko, M. Pooga, R. Brasseur and Ü. Langel, Biochim. Biophys. Acta, 2000, 1467, 165–176 CrossRef CAS.
- (a) A. Elmquist, M. Lindgren, T. Bartfai and Ü. Langel, Exp. Cell Res., 2001, 269, 237–244 CrossRef CAS PubMed; (b) A. Elmquist, M. Hansen and Ü. Langel, Biochim. Biophys. Acta, 2006, 1758, 721–729 CrossRef CAS PubMed.
- (a) H. Brooks, B. Lebleu and E. Vives, Adv. Drug Delivery Rev., 2005, 57, 559–577 CrossRef CAS PubMed; (b) G. M. Poon and J. Gariepy, Biochem. Soc. Trans., 2007, 35, 788–793 CrossRef CAS PubMed; (c) A. Ziegler, Adv. Drug Delivery Rev., 2008, 60, 580–597 CrossRef CAS PubMed.
- S. Futaki, M. Niwa, I. Nakase, A. Tadokoro, Y. Zhang, M. Nagaoka, N. Wakako and Y. Sugiura, Bioconjugate Chem., 2004, 15, 475–481 CrossRef CAS PubMed.
- (a) J. J. Khandare, S. Jayant, A. Singh, P. Chandna, Y. Wang, N. Vorsa and T. Minko, Bioconjugate Chem., 2006, 17, 1464–1472 CrossRef CAS PubMed; (b) P. Zhao and D. Astruc, ChemMedChem, 2012, 7, 952–972 CrossRef CAS PubMed; (c) R. Colombo, M. Mingozzi, L. Belvisi, D. Arosio, U. Piarulli, N. Carenini, P. Perego, N. Zaffaroni, M. De Cesare, V. Castiglioni, E. Scanziani and C. Gennari, J. Med. Chem., 2012, 55, 10460–10474 CrossRef CAS PubMed.
- (a) E. A. Dubikovskaya, S. H. Thorne, T. H. Pillow, C. H. Contag and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12128–12133 CrossRef CAS PubMed; (b) P. A. Wender, W. C. Galliher, N. M. Bhat, T. H. Pillow, M. M. Bieber and N. N. Teng, Gynecol. Oncol., 2012, 126, 118–123 CrossRef CAS PubMed.
- (a) R. S. Navath, Y. E. Kurtoglu, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Bioconjugate Chem., 2008, 19, 2446–2455 CrossRef CAS PubMed; (b) Y. E. Kurtoglu, R. S. Navath, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Biomaterials, 2009, 30, 2112–2121 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis and cellular experiments. See DOI: 10.1039/c4cc02780a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |