 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Label-free, optical sensing of the supramolecular assembly into fibrils of a ditryptophan–DNA hybrid†
Nidhi
Gour‡
a,
Jancy Nixon
Abraham‡
a,
Mohamed
Chami
b,
Axel
Castillo
a,
Sandeep
Verma
c and
Corinne
Vebert-Nardin
*a
aUniversity of Geneva, Faculty of Sciences, Department of Inorganic and Analytical Chemistry, 1211 Geneva 4, Switzerland. E-mail: corinne.vebert@unige.ch; Fax: +41 22 379 60 69; Tel: +41 22 379 64 22
bCenter for Cellular Imaging and NanoAnalytics (C-CINA), Biozentrum, University Basel, Mattenstrasse 26, CH-4058 Basel, Switzerland. E-mail: mohamed.chami@unibas.ch; Fax: +41-61-387 39 86; Tel: +41-61-387 32 24
cDepartment of Chemistry, Centre for Environmental Sciences and Engineering, DST Thematic Unit of Excellence on Soft Nanofabrication, Indian Institute of Technology Kanpur, 208016 (UP), India. E-mail: sverma@iitk.ac.in; Tel: +91-512-259-7643
First published on 13th May 2014
Abstract
The grafting of a short nucleic acid strand to ditryptophan dipeptide (WW) results in a peptide–DNA hybrid, which assembles into fibrils under controlled aggregation conditions as evidenced by label free optical sensing owing to the intrinsic fluorescence of the dipeptide.
The synthesis of amino acid sequences and hybrids thereof is an elegant and versatile approach to assemble nanostructures. Moreover, the functional residues along the amino acid chains infer responsiveness to a variety of physicochemical stimuli. For instance, sensitivity to ionic strength or temperature might induce size variation;1 whereas pH changes trigger the release of an encapsulated compound,2 biological stimulus responsiveness induces morphological transitions.3 Along these lines, the self-assembly of WW-based hybrids has been reported previously.4 The resulting structures were demonstrated to be responsive to various stimuli such as sunlight,5 the presence of potassium ions4b and pH variation.4a Of major interest though is the intrinsic fluorescence of WW and the hybrids thereof, which could be used to monitor changes in the solvent polarity6 as well as specific biotin–avidin and ferrocene–cyclodextrin ligand interactions.7
We recently described the efficient coupling of a short single stranded synthetic nucleotide sequence to the diphenylalanine (FF) dipeptide to induce the formation of pH sensitive vesicular structures.2 In comparison to phenylalanine, tryptophan reveals superior photophysical properties in solution, which are sensitive to the peptide–protein microenvironment.8 Moreover, tryptophan has a stronger electrical quadrupole strength, which allows more favourable stacking of tryptophan containing constructs and assembly of stable structures.9 Tryptophan based peptide networks exhibit superior electron transfer and conductivity properties as well in comparison to phenylalanine based analogues.10 Owing to these peculiar photophysical properties of tryptophan, we thus report herein on the conjugation of WW with a nucleic acid strand to develop functional nanostructures amenable to optical sensing of high potential for diagnostic applications. Of particular interest is the intrinsic fluorescence of ditryptophan, which enables further label-free optical monitoring of structure formation of the resulting peptide DNA hybrid through fluorescence spectroscopy.
Grafting by conventional solid phase synthesis (see ESI†) of the WW dipeptide to a model linear (5′-CTC TCT CTC TTT-3′ 5′-(CT)5T2-3′) synthetic single stranded nucleotide sequence (modified at the 5′ end by an amino group) results in an amphiphilic, rod-coil peptide–DNA hybrid (Scheme 1).
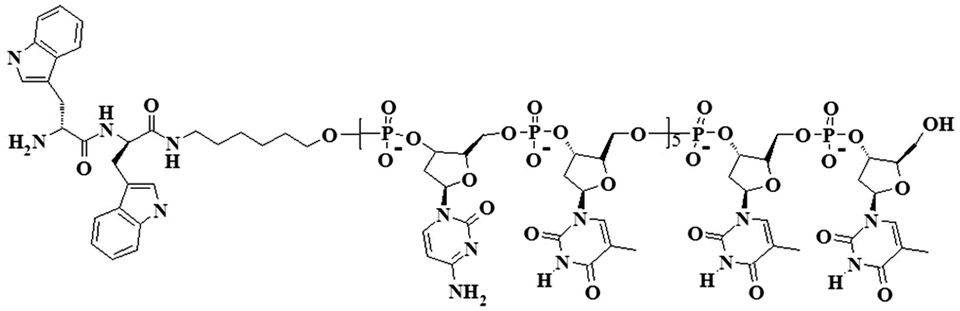 | ||
| Scheme 1 Macromolecular structure of the WW–((CT)5T2) (peptide–DNA) hybrid. | ||
Chemical characterization by HPLC (Fig. S1, ESI†) and ESI-MS (Fig. S2, ESI†) evidences the formation of the peptide–DNA hybrid in accordance with its chemical structure. Owing to both chemical and physical incompatibility between the rigid hydrophobic dipeptide fragment and the water soluble flexible nucleotide sequence, the amphiphilic molecule organizes into spherical structures in dilute aqueous solution (Fig. 1a). Depending on concentration and incubation time, fibrils of a thickness of 0.5–1 μm and length of several micrometres developed (Fig. 1b). As could be observed by transmission and scanning electron microscopy (TEM and SEM), direct dissolution in aqueous solution induced the spontaneous formation of spherical structures of about 300 nm size (Fig. 1a and Fig. S3, ESI†). An apparent hydrodynamic radius (230 nm) of the spherical structures assembled in the low concentration regime was determined by dynamic light scattering (Fig. S4, ESI†), which was in agreement with the size estimated by electron microscopy. The dipeptide itself is not soluble in pure water but spherical structures assembled in an aqueous solution of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v HFIP/H2O ratio) (Fig. 2). Removal of the organic solvent induces the precipitation of the dipeptide. Structure formation in the aqueous solution of HFIP occurs owing to the polar property of the indole ring of tryptophan, which acts as a quadruple as demonstrated in the formation of vesicular structures from N-alkylindoles.9
:50 v/v HFIP/H2O ratio) (Fig. 2). Removal of the organic solvent induces the precipitation of the dipeptide. Structure formation in the aqueous solution of HFIP occurs owing to the polar property of the indole ring of tryptophan, which acts as a quadruple as demonstrated in the formation of vesicular structures from N-alkylindoles.9
 | ||
| Fig. 1 Morphological characterization by scanning electron microscopy of the structures assembled by WW–((CT)5T2): (a) spherical structures assembled at low concentration (below 1 mM) whereas (b) fibres developed at high concentration under controlled aggregation (time and temperature). | ||
 | ||
| Fig. 2 Spherical structures of WW dipeptide in aqueous solution of HFIP as observed by (a) scanning electron microscopy and (b) transmission electron microscopy. | ||
The peptide–DNA hybrid might therefore form the observed fibrils by both π–π stacking and hydrogen bonding according to a nucleation polymerization process comparable to the one that induces the formation of polypeptide fibrils.11 The 230 nm size spherical structures would then act as seeds for this mechanism to take place. As described by Hamley, polypeptide fibril formation occurs by the interaction between the common main chain polypeptide backbone whereas folding is due to specific interactions of the side chains.11 At low concentration, spherical structures formed by WW–DNA is induced by the chemical and physical incompatibility between the dipeptide and nucleic acid strands and stabilized by hydrogen bonding between the nucleotide sequences engaged in structure formation. These spherical structures at higher concentrations and under controlled aggregation (time and temperature) might develop into fibres by nucleation dependent polymerization.
We could thus resort to label-free fluorescence spectroscopy amenable by the WW dipeptide intrinsic fluorescence to evidence this mechanism of structure formation and the putative role of the interactions involved. This peculiar property of WW is sensitive to the polarity of the environment. Since the supramolecular structures are held by non-covalent interactions, they disassemble below a critical aggregation concentration (cac), which was quantified by fluorescence spectroscopy (Fig. 3). Fig. 4 shows the corresponding concentration dependent fluorescence spectra. The dilution course of the fluorescence intensity shows that below 0.1 mM the structures started to disassemble as further evidenced by SEM (Fig. S5, ESI†). The role of hydrogen bonding in stabilization of the spherical structures was further supported by incubation with a chaotropic agent, urea, which also induced their disassembly as imaged by SEM (data not shown) and fluorescence spectroscopy (Fig. S6, ESI†). These experimental results demonstrate that the spherical structures are stabilized by hydrogen bonding, which occurs between the nucleotide sequences engaged in structure formation. This was further supported by the disassembly of the spherical structures by the addition of synthetic nucleic acid strands (either complementary or non-complementary) in the surrounding medium (Fig. S7, ESI†).
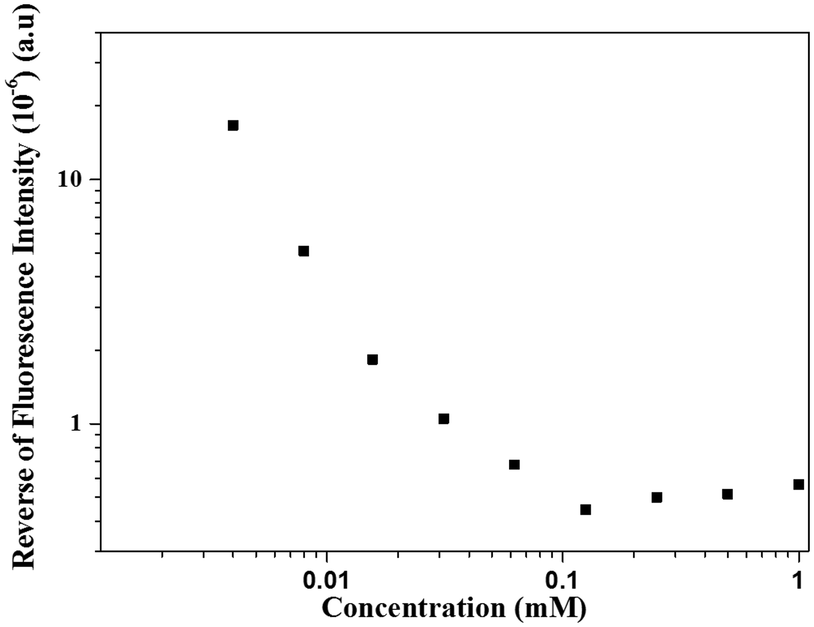 | ||
| Fig. 3 Dilution course of the WW fluorescence intensity. | ||
 | ||
| Fig. 4 Fluorescence emission spectra for decreasing concentrations of the WW–DNA hybrid. (a) 2 mM, (b) 1 mM, (c) 0.5 mM, (d) 0.25 mM, (e) 0.13 mM, and (f) 0.06 mM. | ||
Like dilution, hydrogen bonding between the nucleic acid strands involved in structure formation and the nucleotide sequences in the medium (Fig. S8, ESI†) affects the organization into spherical structures of the WW–DNA. With increasing concentration of the nucleic acid strands, disassembly occurs (Fig. S9, ESI†). Moreover, as can be observed in Fig. 4, the fluorescence intensity maximum (λmax at 370 nm) increases with increasing concentration and the maximum is reached at a concentration of 0.5 mM. On further increasing the concentration (1 mM), the intensity of this peak decreases and a red-shifted signal appears at 476 nm. On further increasing the concentration to 2 mM, this feature becomes more prominent. This red shifted peak is an indication of the presence of an extensive J-type aggregate, which results from the π–π stacking of aromatic rings. These types of aggregates were indeed observed previously in other systems.12 This is further evidenced by SEM (Fig. 1). WW–DNA hybrids organize into spherical structures at concentrations between 0.05 mM and 0.5 mM. On further increasing the concentration (1 mM), they self-assemble to long fibres. These experimental results indicate that structures formed at high concentrations are supported by the interaction of the aromatic WW dipeptide in agreement with reports published previously by Reches and Gazit13 as well as Verma and coworkers.4a We unfortunately could not resort to CD spectroscopy to evidence further π–π stacking. Both the nucleic acid strand and WW–DNA hybrid induce a negative shift at 218 nm (Fig. S10, ESI†).
In addition to π–π stacking of WW residues, the nucleic acid strands might also undergo hydrogen bonding in the fibrillar structure. To assess the role of these interactions, we grafted the WW dipeptide to a water soluble polymer, poly(ethylene glycol) (PEG, MW = 2000). Unlike a nucleotide sequence, PEG does not engage in specific hydrogen bonding. Long needle shaped fibres of about 50 nm width were observed by TEM (Fig. S11, ESI†). Unlike WW–DNA, no spheres were assembled by WW–PEG at low concentration. Similar results were previously reported for PEG-FFFF.12a The authors associated the finite length of the PEG–tetrapeptide hybrid with the end cap energy due to the hydrophobicity of the peptide. WW–PEG thus assembles through β-sheet formation at high concentration due to the π–π stacking of the amino acids which is in agreement with J-type aggregation, as observed for the WW–DNA hybrids through fluorescence spectroscopy (Fig. 4). It further shows that hydrogen bonding plays a key role in the formation of the long WW–DNA fibres. Besides, it has to be emphasized that fibrils could not be assembled by the WW–DNA hybrid in presence of the complementary nucleotide sequence of the one involved in structure formation (data not shown). Moreover, the fibres could not be disassembled by the addition of the complementary sequence subsequent to fibril formation which strongly supports the involvement of hydrogen bonding in WW–DNA fibrils formation in support of π–π stacking. The WW–DNA hybrid organizes according to a mechanism analogous to that of the nucleation polymerization of polypeptide fibrils as further evidenced by the monitoring of the time course of the dipeptide fluorescence (Fig. 5). The peak assigned to the formation of J-type aggregates appears after 6 hours of incubation.
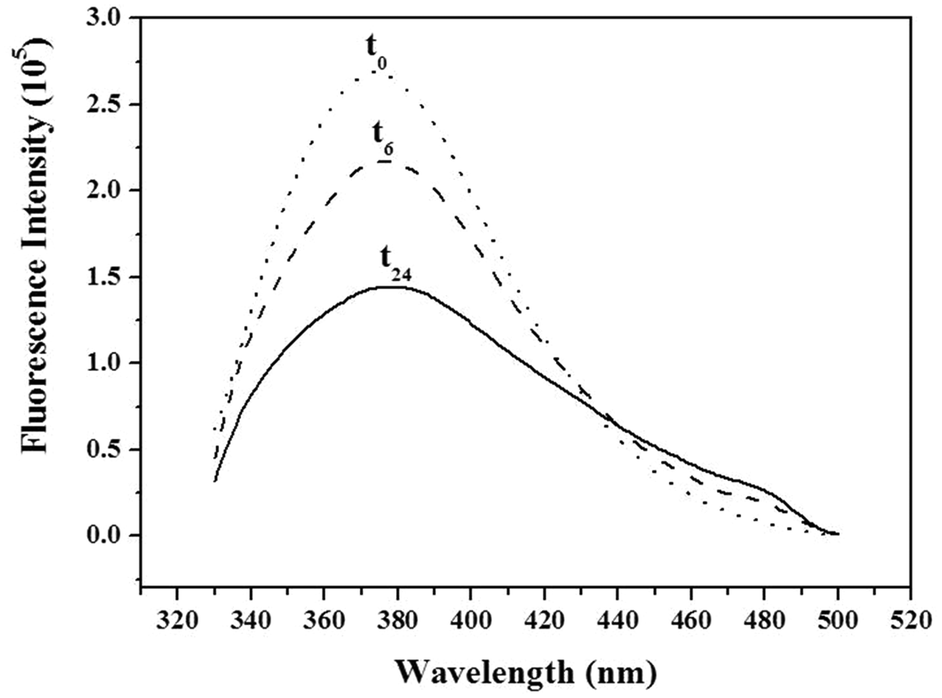 | ||
| Fig. 5 Time course of the fluorescence intensity monitored upon assembly of WW–DNA (0.5 mM) at time intervals 0 (t0), 6 (t6) and 24 hours (t24). | ||
In conclusion, we report here on the successful coupling of the ditryptophan dipeptide to a synthetic nucleotide sequence. The resulting hybrid forms spherical structures in dilute aqueous solution. At high concentration and under controlled aggregation (time and temperature), the formation of fibrils has been observed. Of high interest is the intrinsic fluorescence of the dipeptide which enables the quantification of a critical aggregation concentration and the indirect evidence in a label-free optical mode that the WW–DNA hybrid organization is analogous to that of the nucleation polymerization of polypeptide fibrils, i.e. supported by both main chains π–π stacking of the dipeptide and side chains hydrogen bonding between the nucleic acid strands. This is supported by concentration and time dependent fluorescence spectroscopy and monitoring of the fluorescence spectra of the structures assembled by the WW–DNA hybrid in presence of either the complementary or the non-complementary sequence of the nucleic acid strands involved in the self-assembly.
Dr Apurba K Barman is greatly acknowledged for experimental support. The University of Geneva and the Swiss National Science Foundation for Research (PPOOP2-12838) are acknowledged for financial support.
Notes and references
- T. B. Schuster, D. de Bruyn Ouboter, E. Bordignon, G. Jeschke and W. Meier, Soft Matter, 2010, 6, 5596 RSC.
- N. Gour, D. Kedracki, I. Safir, K. X. Ngo and C. Vebert-Nardin, Chem. Commun., 2012, 48, 5440–5442 RSC.
- T.-H. Ku, M.-P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS PubMed.
- (a) S. Ghosh, M. Reches, E. Gazit and S. Verma, Angew. Chem., 2007, 46, 2002–2004 CrossRef CAS PubMed; (b) K. B. Joshi and S. Verma, Angew. Chem., 2008, 47, 2860–2863 CrossRef CAS PubMed.
- A. K. Barman and S. Verma, Chem. Commun., 2010, 46, 6992–6994 RSC.
- S. Ghosh, P. Singh and S. Verma, Tetrahedron, 2008, 64, 1250–1256 CrossRef CAS PubMed.
- (a) K. B. Joshi and S. Verma, Biophys. Chem., 2009, 140, 129–132 CrossRef CAS PubMed; (b) S. Mondal, S. Ghosh and S. Verma, Tetrahedron Lett., 2010, 51, 856–859 CrossRef CAS PubMed.
- S. Schweizer and J. Reed, Biophys. J., 2008, 95, 3381–3390 CrossRef CAS PubMed.
- E. Abel, M. F. Fedders and G. W. Gokel, J. Am. Chem. Soc., 1995, 117, 1265–1270 CrossRef CAS.
- N. Amdursky, Phys. Chem. Chem. Phys., 2013, 15, 13479–13482 RSC.
- I. W. Hamley, Angew. Chem., 2007, 46, 8128–8147 CAS.
- (a) V. Castelletto and I. W. Hamley, Biophys. Chem., 2009, 141, 169–174 CrossRef CAS PubMed; (b) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemical synthesis and characterization light scattering data and electron microscopy imaging, pH stability. See DOI: 10.1039/c4cc02631d |
| ‡ The two authors contributed equally to this research work. |
| This journal is © The Royal Society of Chemistry 2014 |