 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise synthesis of a Ru4Cd4 coordination cage using inert and labile subcomponents: introduction of redox activity at specific sites†
Alexander J.
Metherell
and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, S3 7HF, UK. E-mail: m.d.ward@shef.ac.uk; Tel: +44 (0)114 2229484
First published on 24th April 2014
Abstract
The kinetically inert mononuclear complex [RuL3](PF6)2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of fac and mer isomers), with three pendant binding sites, reacts with labile Cd(II) ions to complete the assembly of a Ru4Cd4 cubic coordination cage in which reversible redox behaviour has been introduced at the Ru(II) sites.
:3 mixture of fac and mer isomers), with three pendant binding sites, reacts with labile Cd(II) ions to complete the assembly of a Ru4Cd4 cubic coordination cage in which reversible redox behaviour has been introduced at the Ru(II) sites.
The preparation and host–guest chemistry of coordination cages remains a particularly active field in modern supramolecular chemistry,1,2 due a combination of elegant syntheses of new structures by self-assembly methods, as well as the useful functional behaviour – ranging from drug delivery3 to catalysis4 – that can arise from guest inclusion.
Despite recent progress in this field, most cage complexes are based on just two types of component, i.e. one type of metal ion and one type of bridging ligand. This limits the structural and functional and complexity that may be achievable. Given that the metal ions which form the basis of cage assemblies provide both structural information (via their preferences for specific coordination geometries) and possible functionality via properties such as redox activity, magnetism, colour or luminescence,5 efforts directed at assembling heterometallic cages – with control of which metal ions occupy which sites – are surprisingly limited.
So far, mixed-metal cages and related assemblies have been prepared by one of two strategies. The first involves use of unsymmetrical ligands possessing both hard and soft binding sites which will selectively bind to hard and soft metals, respectively.6 The second involves the use of metal ions with different coordination preferences, such as a combination of octahedral and square-planar metal ions whose requirements can each be satisfied at different positions in the cage.7 Both approaches allow the rational design of heterometallic structures with different metal ions at specific sites. However, these methods cannot be applied to cages (or other polynuclear assemblies) in which all metal ions have the same coordination environment, as the necessary differentiation between sites does not exist. Given the extensive family of homoleptic coordination cages that we have studied in recent years,1c with all metal ions in an octahedral tris-chelate coordination environment, we were interested to see if we could develop a route to formation of heterometallic analogues with control over which metal ion occupies which site.
Our strategy involves a combination of kinetically inert [Ru2+] and kinetically labile [Cd2+] metal centres. This allows inert Ru2+ complexes, which are pre-formed vertices of the cage, to be prepared first. These are then combined with labile Cd2+ ions to complete the assembly process, and the inertness of Ru(II) prevents any scrambling of metal ions between different sites. The use of a combination of ‘inert + labile’ components to control assembly of heteronuclear complexes with similar coordination sites is known in other contexts,8 but application of this method to assembly of large cages remains undeveloped.
The cage we have used is a [M8L12]X16 octanuclear ‘cube’ [M = Co, Cd; X = a mono-anion such as BF4−, ClO4− or BPh4−; L is a bis(pyrazolyl-pyridine) bridging ligand; Fig. 1]. We have reported several examples of such cages; all are based on a metal ion at each vertex of an approximate cube with the bridging ligand L spanning each of the twelve edges, giving each metal ion a tris(pyrazolyl-pyridine) coordination environment.9 Importantly, the assembly requires that two of the metal ions (at either end of the long diagonal) have a fac tris-chelate geometry with the three pyridyl donors on one face of the octahedron and the three pyrazolyl donors on the other; whereas the other six metal ions have a mer tris-chelate geometry. With an inversion centre in the cage, this results in molecular S6 symmetry with the C3/S6 axis through the two fac tris-chelate metal centres (Fig. 1).
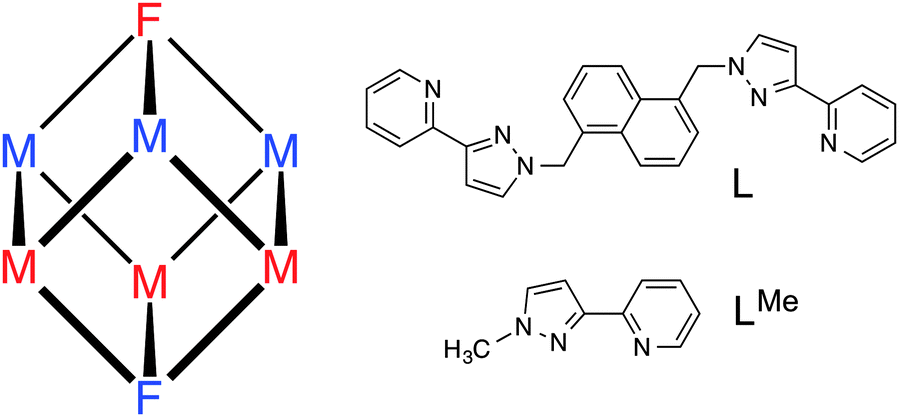 | ||
| Fig. 1 Left: schematic diagram of the cubic cage, showing the positions of the two fac (F) and six mer (M) metal centres. In the homonuclear cages all metal ions are the same; in the heteronuclear cage [Ru4Cd4L12]16+ the two types of metal ion are split over the red and blue sites, with each ion type occupying one fac and three mer centres. Right: the ligand L which connects two metal ions along each edge of the cage, and LMe. | ||
The heterometallic analogue that we report here is [Ru4Cd4L12](ClO4)16 in which the Ru(II) and Cd(II) centres alternate. Each type of metal ion occupies strictly one fac and three mer tris-chelate sites in the cage superstructure. The fac/mer geometric isomerism could add another layer of complexity to the problem of controlled preparation of a heterometallic cage, but in this particular case, it works to our advantage.
Our choice of ‘inert’ metal centre was Ru2+, given its tractable synthetic chemistry: modestly high temperatures suffice for preparation of N,N′-donor tris-chelate complexes but the complexes are generally inert at room temperature. In addition incorporation of Ru2+ centres allows inclusion of a type of functional behaviour (redox activity) that is not normally associated with such cages. If we start with an inert [RuL3]2+ unit as a pre-formed vertex, with three pendant sites at which cage assembly can be propagated by binding to labile Cd2+ ions, it follows that it will not be possible to have two Ru2+ ions adjacent to one another along one edge of the cube. The same is clearly true for the Cd2+ ions given that all available free binding sites are at one end of a bridging ligand whose other terminus is occupied by a Ru2+ ion. The result must be strict alternation of the metal sites around the cube: this can be achieved in two ways which, due to the S6 symmetry of the cube, are degenerate (Fig. 1). Consequently, a 3:1 mixture of mer:fac [RuL3]2+ isomers would provide four pre-formed corners of the cube as the correct isomers, as well as all twelve ligands necessary to complete the assembly. Addition of four equivalents of a labile metal ion that forms octahedral tris-chelate complexes will complete the cube assembly with each type of ion in predictable positions (Fig. 2).
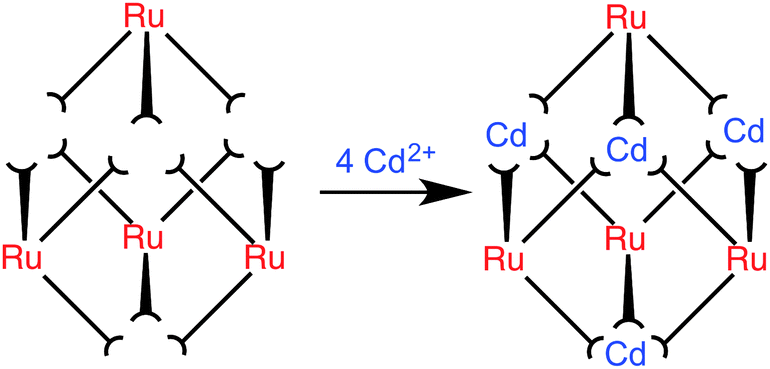 | ||
| Fig. 2 Schematic diagram of the reaction between four pre-formed [RuL3]2+ complex units (each with three pendant binding sites) and four Cd2+ ions to complete assembly of the [Ru4Cd4L12]16+ cage. Arrangement of fac and mer centres is as shown in Fig. 1. | ||
[RuL3](PF6)2 was prepared by reaction of RuCl2(dmso)4 with >3 equiv. L in refluxing ethylene glycol.† Given the non-symmetrical nature of the pyrazolyl-pyridine chelates, of course this forms as a mixture of fac and mer isomers. If there is no specific factor resulting in preference for one isomer over the other, a fac:mer ratio of 1:3 is expected. The 1H NMR spectrum of [RuL3](PF6)2 is consistent with this, showing four independent ligand environments in equal abundance.†‡ In the [RuL3]2+ complex cation, each ligand uses only one of its two chelating sites so there are three pendant pyrazolyl-pyridine binding sites.
Conveniently for our purposes, this 1:3 fac:mer ratio of [RuL3](PF6)2 isomers is precisely what is required in the cage if every alternate site is occupied by a Ru(II) centre. This is not generally true of other members of the cage family, which contains examples in which the metal tris-chelate centres are all fac and other examples in which the metal centres are all mer.1d Thus, no separation of isomers of [RuL3](PF6)2 is needed: the as-prepared mixture can be used as it stands to provide the necessary cage subcomponents in the correct proportions.
The second step was to complete the assembly of the [Ru4Cd4L12]16+ cage by combining [RuL3](PF6)2 with labile Cd2+ ions in a 1:1 ratio, i.e. four of each type of unit as the cage requires (Fig. 2). The twelve pendant bidentate binding sites from four [RuL3]2+ cations are exactly sufficient to combine with four Cd2+ ions (4[RuL3]2+ + 4Cd2+ = [Ru4Cd4L12]16+), and the only way in which cage assembly can be completed is if the Cd2+ and Ru2+ centres are strictly alternating, as shown in Fig. 2.
Reaction of [RuL3](PF6)2 (mix of isomers) with excess of Cd(ClO4)2 (to ensure completion of the assembly) in MeNO2 at RT, followed by diffusion of di-isopropyl ether vapour into the solution, afforded a crop of small orange crystals. X-ray crystallographic analysis§ revealed the structure of the expected octanuclear cage (Fig. 3).9 The key issue is crystallographic location of the Ru2+ and Cd2+ ions at different sites in the cage, which is non-trivial given their similar electron density and size which could lead either to disorder or to mis-identification.
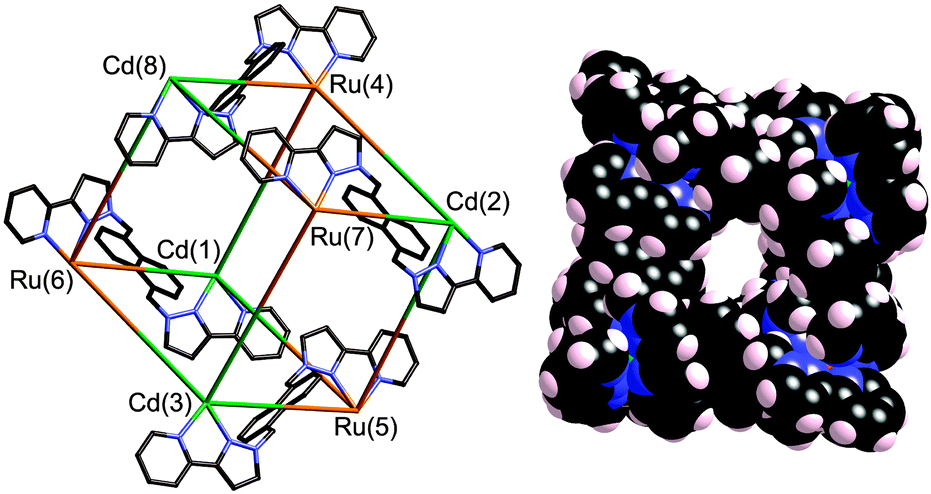 | ||
| Fig. 3 Two views of the cage complex cation in the structure of [Ru4Cd4L12][ClO4]16. Left: a view emphasizing the approximately cubic array of metal ions with four of the bridging ligands includes; right, a space-filling view of the complete cage. | ||
Two distinct pieces of crystallographic evidence confirmed the presence of four Ru2+ and four Cd2+ ions in the desired alternating arrangement. Firstly, these two ions should have different average M–N distances, with Ru–N distances shorter than Cd–N. The four metal positions identified as Ru2+ consistently had significantly shorter bond distances (average, 2.17 Å) than the four positions identified as Cd2+ (average, 2.23 Å).† Secondly, correct assignment of Ru/Cd positions resulted in all eight metal ions having comparable isotropic displacement parameters; inversion of the assignment, i.e. deliberately mis-labelling Ru as Cd and vice versa, resulted in one set of displacement parameters being significantly larger than the other, as expected.
The crystalline product was further analysed by ES mass spectrometry and 1H NMR spectroscopy.† The ES mass spectrum reveals a series of peaks at m/z [Ru4Cd4L12(ClO4)16−z]z+ (z = 4–9) corresponding to the intact complex cation associated with varying numbers of anions (Fig. 4). High-resolution ES spectra give sets of peak clusters for the ions with z = 5, 6, 7 that match exactly what is expected.† A 1H NMR spectrum of [Ru4Cd4L12](ClO4)16 in CD3NO2 was not very informative as it contains 88 independent proton environments in the region 4.7–8.4 ppm;†¶ even at 800 MHz the signals overlap too much for meaningful assignment. However, a DOSY spectrum showed that all of the signals have the same diffusion constant, confirming the presence of a single large assembly in solution.†
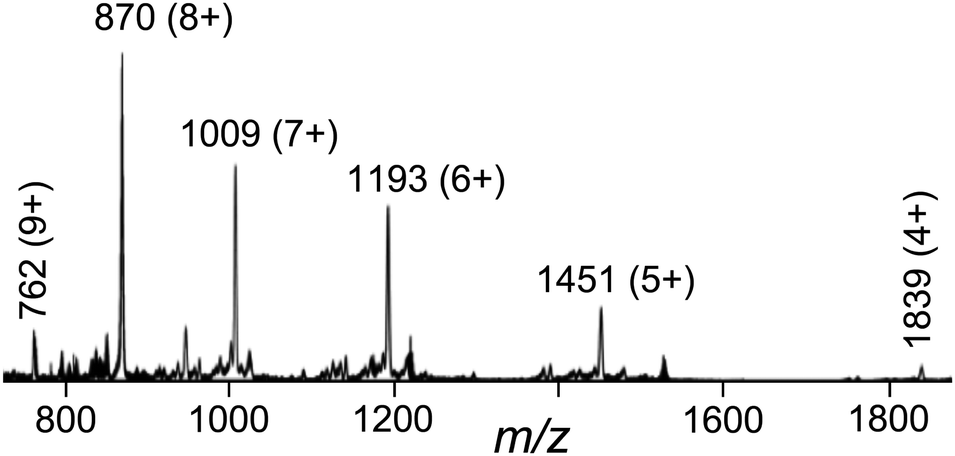 | ||
| Fig. 4 Electrospray mass spectrum of [Ru4Cd8L12][ClO4]16 showing a sequence of peaks corresponding to [Ru4Cd4L12(ClO4)16−z]z+, i.e. loss of 4–9 perchlorate anions from the complete complex. | ||
Finally we investigated the electrochemical behaviour of the cage. The model complex [Ru(LMe)3](PF6)2 (Fig. 1; separate fac and mer isomers)10 shows a reversible Ru2+/Ru3+ wave at +0.85 V vs. ferrocene/ferrocenium (Fc/Fc+) for the fac isomer, and +0.81 V for the mer isomer – a difference of only 40 mV between the isomeric forms. For [Ru4Cd4L12](ClO4)16 we observed a single symmetric wave at +0.96 V vs. Fc/Fc+, which we ascribe to all four Ru2+/Ru3+ couples that are coincident because of the absence of electronic coupling between the Ru centres. The separate processes for the fac and mer centres are also not resolved, but the wave is slightly broadened (ΔEp = 120 mV).†
An important consequence of this redox activity is that the charge on the cage can be switched reversibly between 16+ and 20+. Given that we recently demonstrated how binding of electron-rich organic guests involves a substantial contribution from charge-assisted hydrogen-bonding to the internal surface of the cage, at the position where the electrostatic potential is most positive,9c a reversible redox swing should affect the strength of the host–guest interaction and may provide a mechanism for controlling uptake and release of bound guests. Redox changes also offer the possibility of reversible changes in the luminescence5a,9a or chromic5b properties of the cage.
In conclusion, we have used a combination of kinetically inert and labile metal ions for the rational design and synthesis of a heterometallic Ru4Cd4 coordination cage, in which (i) the four Ru2+ and four Cd2+ ions occupy specific sites in the array; and (ii) we have introduced redox activity associated with the Ru2+ sites.
We thank the EPSRC for financial support, Mr Will Cullen and Dr Andrea Hounslow for recording the 1H NMR spectra, and Mr Harry Adams for assistance with the X-ray crystallography.
Notes and references
- (a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (d) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS PubMed; (f) A. F. Williams, Coord. Chem. Rev., 2011, 255, 2104 CrossRef CAS PubMed; (g) Z. Laughrey and B. Gibb, Chem. Soc. Rev., 2011, 40, 363 RSC; (h) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2012, 254, 1760 CrossRef PubMed; (i) R. J. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (j) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed; (k) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (l) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (m) T. Nakamura, H. Ube and M. Shionoya, Chem. Lett., 2014, 42, 328 CrossRef.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC.
- (a) J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461 CrossRef CAS PubMed; (b) B. Therrien, Chem. – Eur. J., 2013, 19, 8378 CrossRef CAS PubMed; (c) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC.
- (a) C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530 CrossRef CAS PubMed; (b) C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS PubMed; (c) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed.
- (a) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (b) K. Yamashita, M. Kawano and M. Fujita, Chem. Commun., 2007, 4102 RSC.
- (a) W. J. Ramsay, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 13439 CrossRef CAS PubMed; (b) X. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752 CrossRef CAS PubMed; (c) F. E. Hahn, M. Offermann, C. SchulzeIsfort, T. Pape and R. Frohlich, Angew. Chem., Int. Ed., 2008, 47, 6794 CrossRef CAS PubMed; (d) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058 CrossRef CAS PubMed; (e) H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343 CrossRef CAS PubMed; (f) A. J. Metherell and M. D. Ward, RSC Adv., 2013, 3, 14281 RSC.
- (a) V. C. M. Smith and J.-M. Lehn, Chem. Commun., 1996, 2733 RSC; (b) M. M. J. Smulders, A. Jimenez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed; (c) K. Li, L.-Y. Zhang, C. Yan, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed; (d) F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688 CrossRef CAS PubMed; (e) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170 CrossRef CAS PubMed; (f) A. Galstyan, P. J. Sanz Miguel, K. Weise and B. Lippert, Dalton Trans., 2013, 42, 16151 RSC; (g) P. de Wolf, S. L. Heath and J. A. Thomas, Chem. Commun., 2002, 2540 RSC; (h) M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743 CrossRef CAS PubMed.
- (a) Y.-T. Chan, X. Li, J. Yu, G. A. Carri, C. N. Moorefield, G. R. Newkome and C. Wesdemiotis, J. Am. Chem. Soc., 2011, 133, 11967 CrossRef CAS PubMed; (b) H. Sato, A. Nakao and A. Yamagishi, New J. Chem., 2011, 35, 1823 RSC.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC; (c) S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed; (d) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC.
- A. J. Metherell, W. Cullen, A. Stephenson, C. A. Hunter and M. D. Ward, Dalton Trans., 2014, 43, 71 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF format; bond distances and angles around the metal ions; further details of the crystallographic refinement; details of synthesis and characterisation; details of NMR and mass spectrometric characterisation of the cage; cyclic voltammograms. CCDC 996081. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02627f |
| ‡ The mer isomer has no symmetry with all three ligands inequivalent; the fac isomer provides the fourth ligand environment with all three ligands equivalent due to the threefold symmetry but is only one-third as abundant. Hence we see signals for four independent ligand environments in equal abundance. |
| § Crystal data for [Ru4Cd4L12](ClO4)16·{[Cd(H2O)6](ClO4)}0.5·11MeNO2·3H2O: C347H309Cd4.5Cl17N83O96Ru4, M = 8441.94 g mol−1, monoclinic, space group P21/c, a = 23.0027(14), b = 40.888(2), c = 50.529(3) Å, β = 100.989(3)°, U = 46653(5) Å3, Z = 4, T = 100(2) K, λ(Mo-Kα) = 0.71073 Å. 325928 reflections were collected (2θmax = 45°) which after merging afforded 61008 independent reflections with Rint = 0.176. Final R1[I > 2σ(I)] = 0.130; wR2 (all data) = 0.378. See ESI† for further details. |
| ¶ The homonuclear cages [M8L12]X16 contain 44 proton environments because the two different ligand environments (connecting fac/mer and mer/mer metal centres, with six ligands in each environment) have no internal symmetry (ref. 9). In the Ru4Cd4 complex the symmetry is reduced by a further factor of two due to loss of the inversion centre. |
| This journal is © The Royal Society of Chemistry 2014 |