 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Broadening the scope of Baeyer–Villiger monooxygenase activities toward α,β-unsaturated ketones: a promising route to chiral enol-lactones and ene-lactones†
T.
Reignier‡
a,
V.
de Berardinis‡
b,
J.-L.
Petit
b,
A.
Mariage
b,
K.
Hamzé
a,
K.
Duquesne
a and
V.
Alphand
*a
aAix Marseille Université, Centrale Marseille, CNRS iSm2 UMR 7313, 13397 Marseille, France. E-mail: v.alphand@univ-amu.fr; Fax: +33 4 91 28 84 40
bCEA, Institut de Genomique, Genoscope, Université Evry Val d'Essonne (UEVE), CNRS, UMR Génomique métabolique, 2 rue Gaston Crémieux, 91057 Evry, France
First published on 22nd May 2014
Abstract
Three regiodivergent Baeyer–Villiger mono-oxygenases (enantioselectively) oxidized a series of cyclic α,β-unsaturated ketones into (chiral) either enol-lactones or ene-lactones. An easy-to-use and efficient biocatalytic process based on a host-microorganism deprived of unwanted activities (knock-out mutant) was developed to enable the exclusive synthesis of unsaturated lactones.
Conjugated ene-lactones1 and enol-lactones2 are frequently used as motifs in diverse bioactive synthetic and natural products. They are also valuable intermediates in organic synthesis3 and offer a promising route to the synthesis of other motifs widely found in nature such as dihydrooxepines.4 Nevertheless, the synthesis of such lactones, especially in the context of medium size rings, is not straightforward.5,6 The classical ring-closing approaches such as lactonization1f,2d and ring-closing metathesis7 can be inefficient due to kinetic or thermodynamic limitations.5 The most direct route, the Baeyer–Villiger (BV) oxidation of α,β-unsaturated ketones, when performed chemically always produces racemic enol-lactone and suffers from frequent side-reactions.8 Regarding enzymatic BV oxidation it is commonly thought that enones are not oxidized by Baeyer–Villiger monooxygenases (BVMOs) even though these enzymes display both regioselectivity and high stereospecificity toward a large range of saturated ketones.9
BVMOs are a highly versatile class of flavoenzymes able to perform the efficient catalysis of chemo-, regio- and enantioselective oxygenation reactions.9 Classically, an atom from dioxygen is incorporated to transform ketones into esters or lactones while consuming NADPH, a cofactor required as an electron donor. The reactive species, C4a-peroxyflavin,10 acts as a nucleophile to give a Criegee-like intermediate that undergoes the same type of rearrangement as in chemical BV reactions. Besides BV oxidation of ketones, the enzymes are able to oxygenate sulfur, selenium, nitrogen, boron atoms and, much more exceptionally, epoxidize the double bond. The main difference when compared to chemical reagents comes from the general lack of activity against α,β-unsaturated ketones.9,11 To the best of our knowledge, the only unambiguous mention of such a reaction being enzymatically catalyzed was reported in 1996: the oxidation of 5-hexyl-2-cyclopenten-1-one by pure cyclopentanone monooxygenase from Comamonas sp. NCIMB 9872 (CPMOComa).12 Since no other publications have described BVMO-mediated enol or ene-lactone formation,13 thus syntheses of these compounds remain challenging.5,6a
We confirmed here that (asymmetric) enzymatic BV oxidation of cyclic enones is possible and offers a promising route for the synthesis of (chiral) unsaturated lactones. We report for the first time the BV oxidation of a series of α,β-unsaturated ketones using CPMOComa and two new BVMOs. The enzymes displayed a complementary regioselectivity and enantioselectivity, leading to either the corresponding conjugated ene-lactones or enol-lactones. An easy-to-use and efficient biocatalytic process based on a host-microorganism deprived of unwanted endogenous reductase activity was also developed.
BVMOOcean, from Oceanicola batsensis DSM 15984, and BVMOParvi, from Parvibaculum lavamentivorans DSM 13023, were selected in the course of screening of sixty putative bacterial Type I BVMOs chosen from genomic databases to cover the genomic diversity as well as possible.14 The genes were heterologously expressed in E. coli strain BL21(DE3).15 The activities of the corresponding enzymes were assayed against various ketone substrates by monitoring NADPH depletion in crude extracts. Only two extracts displayed activity against cycloalkenones 1a–c (Scheme 1), these results were confirmed by experiments on the purified enzymes (see ESI†).
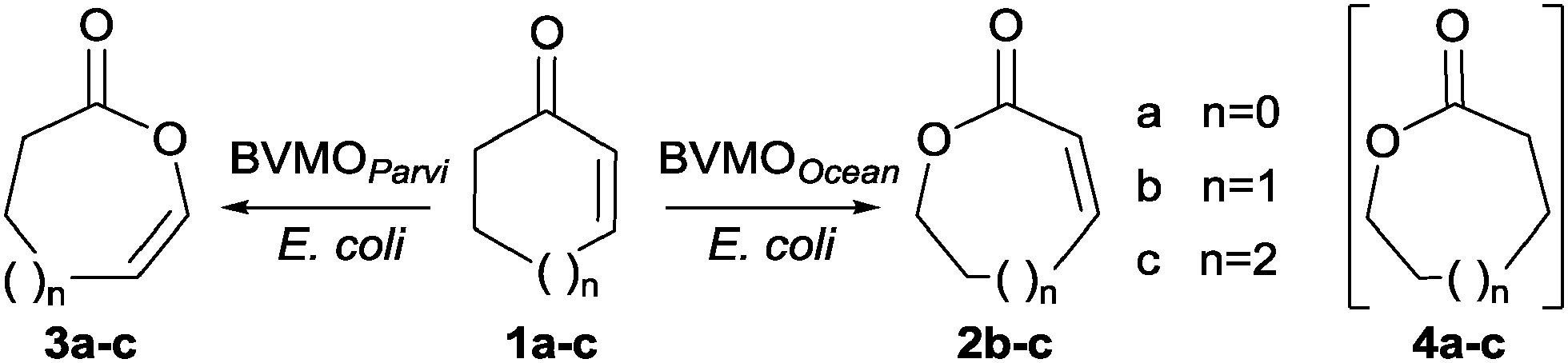 | ||
| Scheme 1 Biotransformations using BVMOOcean and BVMOParvi expressed in E. coli BL21(DE3) strains. | ||
Whole-cell biotransformations16 of 1a–c were carried out for product identification. Unsaturated lactones were formed with both strains as reported in Table 1. In the experiments with E. coli BL21(DE3) containing BVMOOcean, oxygen insertion took place between the carbonyl group and the non-ethylenic carbon atom to give conjugated ene-lactones 2b and 2c while lactone 2a formation was not observed. The regioselectivity was similar to that previously reported with CPMOComa on 5-hexyl-2-cyclopentenone12 and opposite to that described in the classical chemical BV reaction.8
| Enzymea,b | 1 | Residual 1 yieldc (%) | 2 yieldc (%) | 3 yieldc (%) | 4 yieldc (%) |
|---|---|---|---|---|---|
| a Values corresponding to the experiments carried out with engineered E. coli BL21(DE3) strains are shown in parentheses. b Biotransformations were carried out at the 3 mM scale in 2L flasks. c Yields were determined by GC analysis using decane or undecane as internal standard. d 50% of cycloheptanone was concurrently formed from 1c reduction. | |||||
| BVMOOcean | 1a | 70 (81) | — | — | 8 (0) |
| 1b | — | 39 (79) | — | 43 (5) | |
| 1c | — | 83 (80) | — | 8 (0) | |
| BVMOParvi | 1a | — | — | 81 (85) | 1 (0) |
| 1b | — | — | 60 (74) | 11 (<1) | |
| 1c | 15d (40) | — | 10 (19) | 3 (0) | |
Enol-lactones 3a–c were exclusively produced with E. coli BL21(DE3) containing BVMOParvi. They resulted from the oxygen atom insertion between the carbonyl group and the double bond as observed in chemical BV oxidation. However the epoxidation of the double bond, a frequent side-reaction that has prevented chemical BV oxidation being used for the synthesis of ene-lactones 3a–c,4,8 was not observed.
Construction of an engineered strain deprived of ene-reductase activity: large amounts of saturated ketones or corresponding saturated lactones were formed with both strains (Table 1), lessening the interest in these microbiological transformations. The presence of cycloalkenone reductase activity in E. coli BL21(DE3) was confirmed by whole-cell biotransformation of 1a–c (see ESI†) while unsaturated lactones were not hydrogenated. The reductase required nicotinamide cofactors, with a preference for NADPH as type I BVMOs. The literature11,17 suggested that NemA reductase is a good candidate and its contribution to cycloalkenone hydrogenation was confirmed when we tested the knock-out mutant BW25113ΔnemA obtained from the Keio collection.18 An E. coli BL21(DE3) expression strain without active enone reductase was engineered by exchanging nemA gene for ΔnemA knockout cassette from BW25113ΔnemA using bacteriophage P1 transduction.19 The newly engineered BL21(DE3)ΔnemA strain was then used as a host for the overexpression of BVMOOcean and BVMOParvi.
Preparative biotransformations using newly constructed strains: biotransformations carried out with the new biocatalysts, BL21(DE3)ΔnemA strains expressing BVMOOcean and BVMOParvi, showed the almost complete abolition of the saturated lactone formation (Table 1). These experiments clearly demonstrated that the knock-out strains producing BVMOs were suitable for large scale BV oxidation of cycloalkenones.20
Widening the range of substrates: whole cell biotransformations of methyl substituted cycloalkanones 5a–c and cycloalkenones 5d–g were performed (Scheme 2). All saturated ketones 5a–c were transformed by both enzymes but a strong disparity, depending on substrate and enzyme, was observed in enantio- or enantiotoposelectivity as shown in Tables 2 and 3. The results obtained with BVMOOcean were very close to those reported with the well-known CHMOAcineto, this was consistent with their high sequence identity (58%). However, even though they have an equivalent identity (53%), BVMOParvi and CPMOComa differed in enantioselectivity (see ESI†).
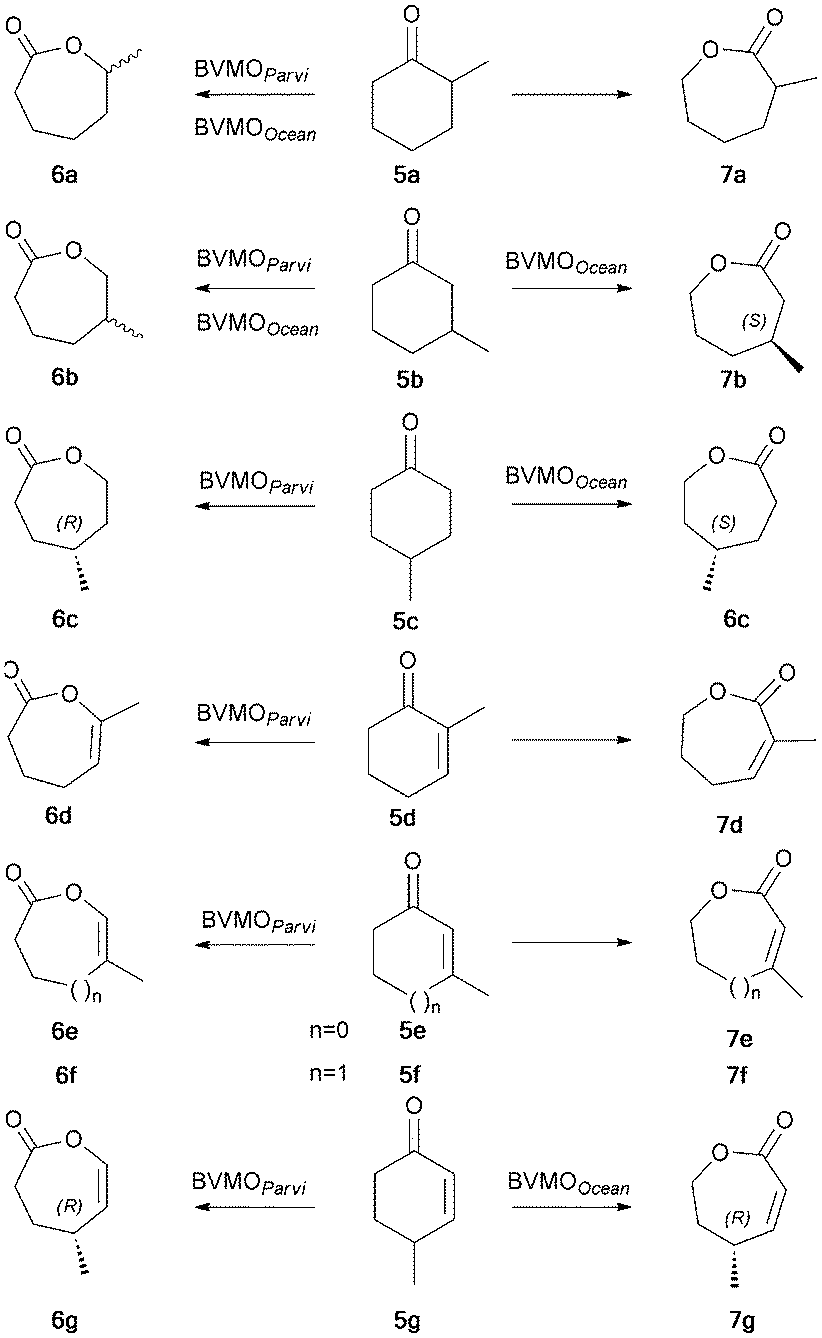 | ||
| Scheme 2 Biotransformation of methylated cycloalkanones and cycloalkenones by BVMOOcean and BVMOParvi expressed in knock-out E. coli strains. | ||
| Ketonea | Residual 5 yieldb (%) | Lactone 6 yieldb (%) | Enantiomeric ratio Ec |
|---|---|---|---|
| ee (abs conf) | ee (abs conf) | ||
| a Preparative biotransformations were carried out at the 3 mM scale in 2L flasks. b Yields were determined by GC analysis using internal standard. c Enantiomeric ratios were calculated from three couples of substrate and product ees. | |||
| 5a | 78 | 21 | |
| 14 ee (S) | 23 ee (R) | 3 | |
| 5b | 79 | 20 | |
| 33 ee (R) | 88 ee (S) | 15 | |
| 5c | — | 93 | |
| 68 ee (R) | — | ||
| 5d | 0 | 71 | — |
| 5e | 0 | 75 | — |
| 5f | 0 | 70 | — |
| 5g | 80 | 15 | |
| 30 ee (S) | 93 ee (R) | 37 | |
| Ketonea | Residual 5 yieldb (%) | Lactone 6 yieldb (%) | Lactone 7 yieldb (%) | E |
|---|---|---|---|---|
| ee (abs conf) | ee (abs conf) | ee (abs conf) | ||
| a Preparative biotransformations were carried out at 3 mM scale into 2L flasks. b Yields were determined by GC analysis using internal standard. c E: enantiomeric ratio, calculated from three couples of substrate and product ees. | ||||
| 5a | 70 | 24 | 0 | |
| 29 ee (R) | 56 ee (S) | — | 5 | |
| 5b | 0 | 50 | 48 | |
| — | >98 ee (R) | >98 ee (S) | — | |
| 5c | 0 | 92 | 0 | |
| — | >98 ee (S) | — | — | |
| 5g | 60 | 0 | 35 | |
| 65 ee (S) | — | 88 ee (R) | 31 | |
The most surprising behavior arose from the reactivity of both new enzymes with substituted cycloalkenones. Only BVMOParvi was able to transform ketones 5d–f and afford exclusively enol-lactones 6d–f in good yields (Table 2) as for experiments with 1a–c. Moreover, highly optically active enol-lactone (R)-6g was obtained from 5g (enantiomeric ratio E = 37), highlighting for the first time the BVMO capacity to catalyze enantioselective enol-lactone formation. On the other hand, 5g was the unique methylated enone of the series to act as a substrate of BVMOOcean, suggesting a strong sensitivity of the enzyme towards the double bond substitution. 5g was oxidized into the conjugated ene-lactone 7g with the same regioselectivity as the non-substituted enones. A high enantioselectivity was also observed (E = 31), leading to the preferential formation of the (R)-enantiomer in good yield (Table 3).
The same experiments were performed with a similarly constructed knock-out E. coli strain producing CPMOComa and revealed a behavior of this enzyme identical to that of BVMOParvi (see Table 4 and ESI†). This outcome highlighted the advantage of our knock-out E. coli strain since in a previous published study based on an unmodified strain, any eventual BV activity of CPMOComa on cyclohexenones was totally masked by reductase activity.21 In contrast, we confirmed, as previously suggested,11 that CHMOAcineto was unable to use cyclohexenones as substrates although sharing 58% sequence identity with BVMOOcean.
Thus the three enzymes displayed the same enantiopreference but a regiodivergence was observed between BVMOOcean on one hand and CPMOComa and BVMOParvi on the other hand. The protein sequences show a low similarity (35–40%) between these two groups, it is likely that their particular activities towards enones come from a very subtle variation in the aminoacid arrangement that will require comparisons with a larger number of enzymes with similar activities before being understood.
In conclusion, a long standing gap in the chemistry of BVMOs has been filled. We confirmed that this family of enzymes is able to convert without exception the same type of compounds as peracids do. The two original activities discovered on cyclic α,β-unsaturated ketones associated with a strategy based on knockout mutant strains allowed easy access to enol-lactones and conjugated ene-lactones, expanding the toolbox of the synthetic chemist. The range of substrates still remains to be explored more widely and the molecular reasons for the rareness of BVMO mediated reactivity towards enones need to be understood. Nevertheless, these preliminary studies, particularly as far as enantioselectivity is concerned, pave the way toward valuable new enantiopure unsaturated synthons.
This work and T. Reignier PhD fellowships were supported by the French National Research Agency (NaturaDyRe project ANR-10-CD2I-014). We are deeply grateful to Dr E. Bouveret, L. My and Dr P. Moreau for biological materials gift and advice, E. Courvoisier-Dezord and Prof. G. Iacazio for AVB Platform facilities and Prof. J. N. Sturgis for his critical reading.
Notes and references
- (a) A. Parenty, X. Moreau, G. Niel and J.-M. Campagne, Chem. Rev., 2013, 113, PR1 CrossRef CAS PubMed; (b) B. Nagaiah and A. V. Narsaiah, Helv. Chim. Acta, 2013, 96, 1948 CrossRef CAS; (c) R. S. Ghogare, S. B. Wadavrao and A. V. Narsaiah, Tetrahedron Lett., 2013, 54, 5674 CrossRef CAS; (d) Y. Zhang, J.-S. Wang, D.-D. Wei, Y.-C. Gu, X.-B. Wang and L.-Y. Kong, J. Nat. Prod., 2013, 76, 1191 CrossRef CAS PubMed; (e) S.-M. Lee, W.-G. Lee, Y.-C. Kim and H. Ko, Bioorg. Med. Chem. Lett., 2011, 21, 5726 CrossRef CAS PubMed; (f) D. K. Reddy, V. Shekhar, P. Prabhakar, B. C. Babu, B. Siddhardha, U. S. N. Murthy and Y. Venkateswarlu, Eur. J. Med. Chem., 2010, 45, 4657 CrossRef CAS PubMed; (g) M. Herrera-Ruiz, M. González-Cortazar, E. Jiménez-Ferrer, A. Zamilpa, L. Álvarez, G. Ramírez and J. Tortoriello, J. Nat. Prod., 2006, 69, 59 CrossRef CAS PubMed; (h) H. Shigemori, S. Shimamoto, M. Sekiguchi, A. Ohsaki and J. Kobayashi, J. Nat. Prod., 2002, 65, 82 CrossRef CAS PubMed; (i) S. V. Ley, L. R. Cox and G. Meek, Chem. Rev., 1996, 96, 423 CrossRef CAS PubMed.
- (a) J. Mo, R. Yang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 50 CrossRef CAS PubMed; (b) R. Larsson, O. Sterner and M. Johansson, Org. Lett., 2009, 11, 657 CrossRef CAS PubMed; (c) J. J. Rubal, F. J. Moreno-Dorado, F. M. Guerra, Z. D. Jorge, M. del Carmen Galán, G. M. Salido, S. B. Christensen, H. Søhoel and G. M. Massanet, Planta Med., 2010, 76, 284 CrossRef CAS PubMed; (d) M. Jiménez-Tenorio, M. C. Puerta, P. Valerga, F. J. Moreno-Dorado, F. M. Guerra and G. M. Massanet, Chem. Commun., 2001, 2324 RSC.
- (a) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 286 CrossRef CAS PubMed; (b) A. Yanagisawa, T. Fujinami, Y. Oyokawa, T. Sugita and K. Yoshida, Org. Lett., 2012, 14, 2434 CrossRef CAS PubMed; (c) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796 CrossRef CAS PubMed; (d) H. Yanai, A. Takahashi and T. Taguchi, Tetrahedron, 2007, 63, 12149 CrossRef CAS; (e) C. D. Hopkins, L. Guan and H. C. Malinakova, J. Org. Chem., 2005, 70, 6848 CrossRef CAS PubMed; (f) K. R. Campos, J. C. S. Woo, S. Lee and R. D. Tillyer, Org. Lett., 2004, 6, 79 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Yu, L. Shi, Q. Cai, M. Lu and P. Heretsch, Org. Lett., 2013, 15, 1994 CrossRef CAS PubMed and references herein.
- W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680 CrossRef CAS PubMed.
- (a) Y. Li, Z. Yu and H. Alper, Org. Lett., 2007, 9, 1647 CrossRef CAS PubMed; (b) A. T. Davies, J. E. Taylor, J. Douglas, C. J. Collett, L. C. Morrill, C. Fallan, A. M. Z. Slawin, G. Churchill and A. D. Smith, J. Org. Chem., 2013, 78, 9243 CrossRef CAS PubMed; (c) X. Hao, X. Liu, W. Li, F. Tan, Y. Chu, X. Zhao, L. Lin and X. Feng, Org. Lett., 2014, 16, 134 CrossRef CAS PubMed.
- A. K. Chatterjee, J. P. Morgan, M. Scholl and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 3783 CrossRef CAS.
- (a) G. R. Krow, Org. React., 1993, 43, 251–798 CAS; (b) C. Jimenez-Sanchidrian and J. R. Ruiz, Tetrahedron, 2008, 64, 2011 CrossRef CAS.
- For recent reviews, see: (a) K. Balke, M. Kadow, H. Mallin, S. Saß and U. T. Bornscheuer, Org. Biomol. Chem., 2012, 10, 6249 RSC; (b) H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165 CrossRef CAS PubMed; (c) V. Alphand and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1928 CrossRef CAS; (d) D. E. Torres Pazmiño, H. M. Dudek and M. W. Fraaije, Curr. Opin. Chem. Biol., 2010, 14, 138 CrossRef PubMed; (e) G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208 CrossRef CAS PubMed.
- (a) B. J. Yachnin, T. Sprules, M. B. McEvoy, P. C. K. Lau and A. M. Berghuis, J. Am. Chem. Soc., 2012, 134, 7788 CrossRef CAS PubMed; (b) D. Sheng, D. P. Ballou and V. Massey, Biochemistry, 2001, 40, 11156 CrossRef CAS PubMed.
- For instance, CHMOAcineto, although known for its outstanding substrate acceptance, did not oxidize cyclohexenones ( N. Oberleitner, C. Peters, J. Muschiol, M. Kadow, S. Saß, T. Bayer, P. Schaaf, N. Iqbal, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2013, 5, 3524 CrossRef CAS ).
- M. T. Bes, R. Villa, S. M. Roberts, P. W. H. Wan and A. Willetts, J. Mol. Catal. B: Enzym., 1996, 1, 127 CrossRef CAS.
- It was recently assumed that OTEMO, a BVMO from Pseudomonas putida, used a few enones as substrates. Unfortunately, no evidence of unsaturated lactone formation has been provided to date ( M. Kadow, K. Loschinski, S. Saß, M. Schmidt and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2012, 96, 419 CrossRef CAS PubMed ).
- 28 sequences coding for experimentally confirmed BVMOs were collected and used for similarity sequence analyses using UniprotKB and Genoscope databases. Candidate enzymes were then selected according to their genome availability in the Genoscope strain collection (700 prokaryote species). Around 370 candidate enzymes were selected after clustering and 180 were cloned (46% of the selected genes had a GC content >65%). Sixty enzymes were overexpressed in E. coli BL21. The whole screening will be detailed elsewhere.
- C. Vergne-Vaxelaire, F. Bordier, A. Fossey, M. Besnard-Gonnet, A. Debard, A. Mariage, V. Pellouin, A. Perret, J.-L. Petit, M. Stam, M. Salanoubat, J. Weissenbach, V. de Berardinis and A. Zaparucha, Adv. Synth. Catal., 2013, 355, 1763 CrossRef CAS.
- In whole-cell processes, cofactor regeneration was provided by the cellular machinery itself and avoided the need for the exogenous NADPH recycling system ( I. Hilker, M. C. Gutiérrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3, 546 CrossRef CAS PubMed ).
- R. E. Williams, D. A. Rathbone, N. S. Scrutton and N. C. Bruce, Appl. Environ. Microbiol., 2004, 70, 3566 CrossRef CAS PubMed.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2 CrossRef PubMed , 2006.0008.
- J. H. Miller, A Short Course in Bacterial Genetics, Cold Spring Harbor Laboratory Press, Plainview, NY, 1992 Search PubMed.
- Moreover, the use of these knock-out strains for future targeting enones screenings could enhance the sensitivity of the assays by decreasing the “background activity” due to NADPH consumption by NemA.
- M. D. Mihovilovic, R. Snajdrova and B. Grötzl, J. Mol. Catal. B: Enzym., 2006, 39, 135 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Enzyme sequences, genomic constructions experimental procedures and product characterization data. See DOI: 10.1039/c4cc02541e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |