 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise synthesis of a stable diphosphasilirane and its unexpected dimerization†
Kirsten
Reuter
and
Carsten
von Hänisch
*
Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35043 Marburg, Germany. E-mail: haenisch@chemie.uni-marburg.de; Fax: +49-6421-2825653
First published on 5th June 2014
Abstract
The synthesis of the thermally stable bicyclic diphosphasilirane O(SiiPr2P)2SiiPr2 (4) was achieved via oxidation of the alkaline earth metal substituted cyclic diphosphanide 3 with 1,2-dibromoethane. Nonetheless, during extended storage at low temperature, impurity induced rearrangement in favour of the dimeric species [O(SiiPr2P)2SiiPr2]2 (5) is observed.
The stability of three-membered heterocycles strongly depends on the skeleton atoms and their substituents. For example the ring strain drops in the series from cyclotrisilane to the heterocyclic compound Si2PH51 and finally to the almost unstrained cyclotriphosphine.2 Also bulky substituents give rise to increased stability of small ring systems.3,4 This can best be illustrated by the ring compound (tBuP)2SiMe2, synthesized by Baudler et al., which easily decomposes in favour of larger ring systems, while in the case of the analogous phenyl substituted (tBuP)2SiPh2 no by-products occur.5
In prior work, alkaline earth metal derivatives of primary and secondary phosphides turned out to be valuable starting materials for oxidative phosphorus–phosphorus coupling.6 With the aim to synthesize a stable diphosphasilirane, we tried to integrate the P2-unit into a robust five-membered ring. Therefore, the cyclic compound 1 seems to be a promising starting material. The synthesis of 1 is accomplished through metalation of O(SiiPr2PH2)2 by nBuLi and subsequent reaction with SiiPr2Cl2 (Scheme 1), which yields 75.3% of a highly-viscous, colourless oil.
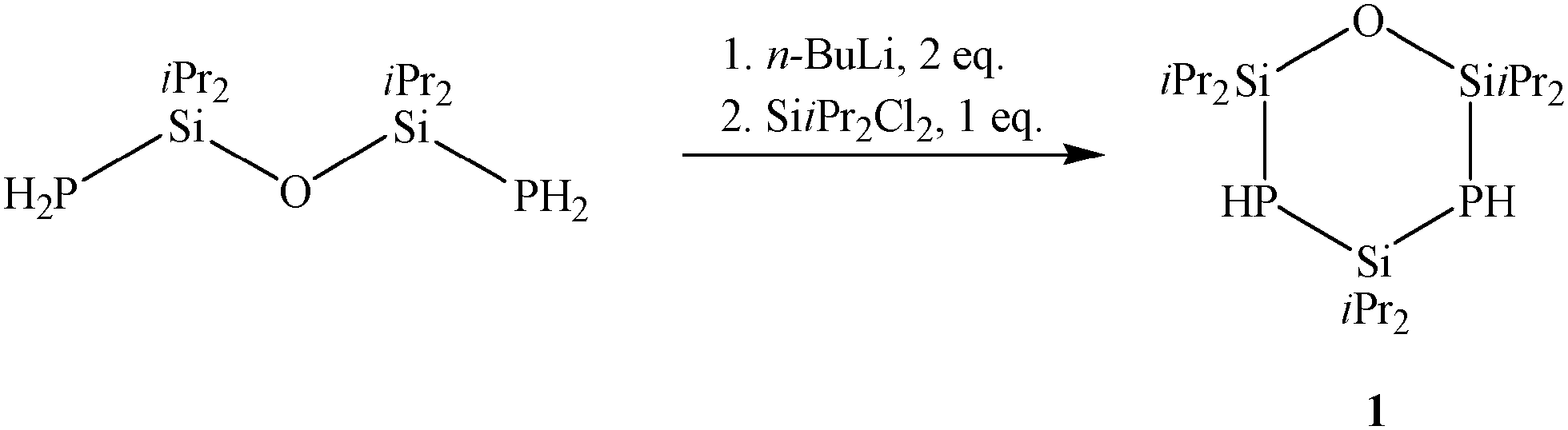 | ||
| Scheme 1 Synthesis pathway of compound 1. | ||
At −25 °C, compound 1 forms colourless crystals which melt at room temperature. In the IR spectrum, the P–H vibration can be observed at 2286 cm−1. Furthermore, according to the equivalent phosphorus atoms, the 31P{1H} NMR spectrum shows one signal at −273.1 ppm, which is broader at room-temperature. Pyramidal inversion is a well-known phenomenon for phosphines.7 As the temperature increases to 77 °C, one sharp signal can be observed at −273.3 ppm (Fig. 1). At −75 °C, the 31P{1H} NMR spectrum displays separate signals at −271.0 and −278.3 ppm which are assigned to the rac and the meso forms. The coalescence temperature amounts to 17 °C, the barrier for pyramidal inversion, calculated by Eyring equation, yields ΔG≠ = 51.0 kJ mol−1.
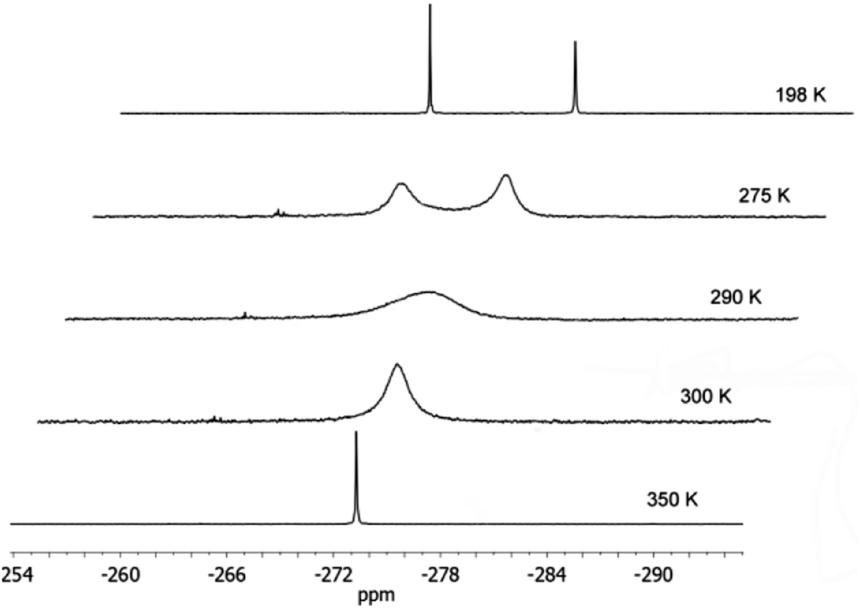 | ||
| Fig. 1 31P{1H} NMR of compound 1. | ||
The reaction of O(SiiPr2PH)2SiiPr2 (1) with the hexamethyldisilazanides of strontium and barium leads to the monomeric products 2 und 3 as shown in Scheme 2. In DME, the colourless complex [Sr{O(SiiPr2P)2SiiPr2}(dme)2] (2) crystallizes in the orthorhombic space group Pbca with eight molecules per unit cell. The dianionic [O(SiiPr2P)2SiiPr2]2− entity acts as a bidentate ligand, coordinating to the strontium cation (Fig. 2). Due to saturation by two DME molecules, the strontium cation achieves the coordination number six. The siloxane group stands in one plane with both phosphorus atoms, while the silane entity has a highly distorted position. Since the Si–P–Si angles amounts to 96.89(2)° and 96.52(1)°, the ligand assumes envelope conformation.
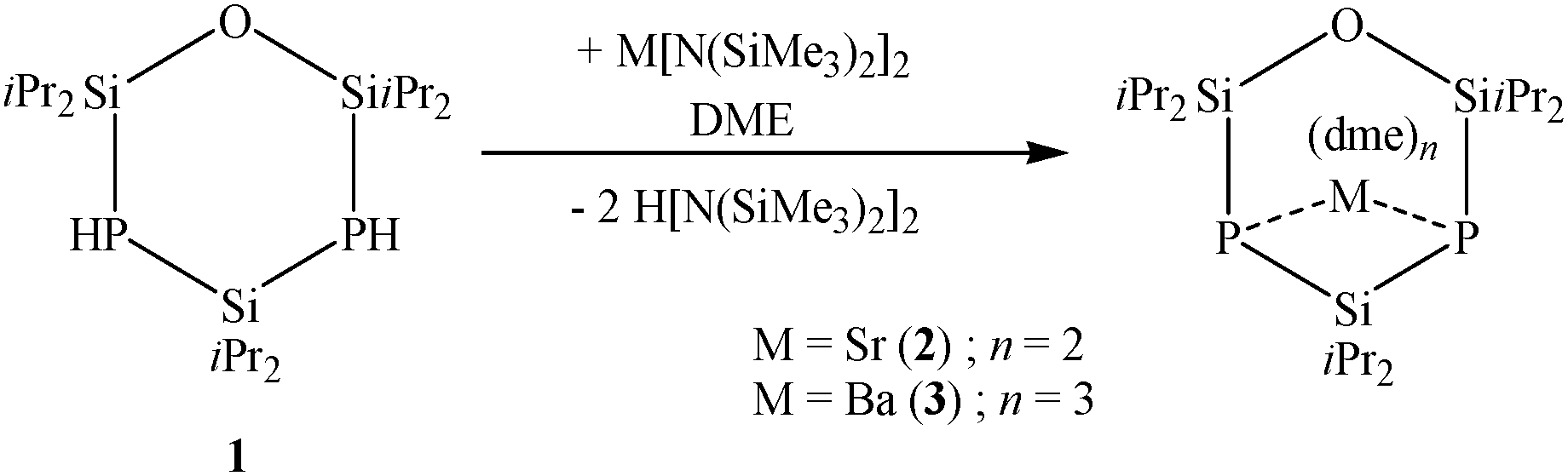 | ||
| Scheme 2 Reaction of 1 with disilazanides of the heavy alkaline earth metals. | ||
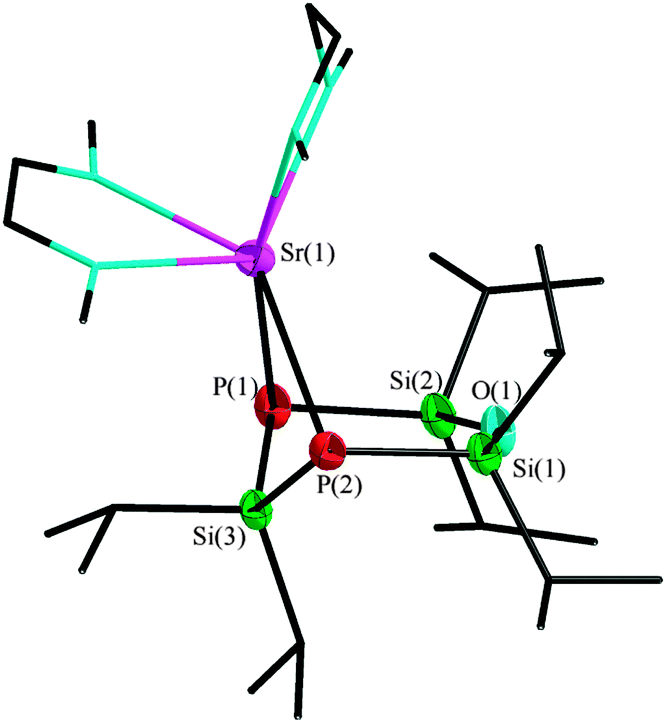 | ||
| Fig. 2 Molecular structure of 2, thermal ellipsoids represent a 50% probability level, hydrogen atoms of the organic groups are not shown. Selected atom distances [pm] and angles [°]: P(1)–Sr(1) 305.9(1), P(2)–Sr(1) 303.3(1), P(1)–Si(3) 224.6(1), P(2)–Si(3) 224.5(1), P(1)–Si(1) 220.2(1), P(1)–Si(2) 220.8(1), Si(1)–O(1) 165.7(0), Si(2)–O(1) 165.1(1); P(1)–Sr(1)–P(2) 76.19(1), P(1)–Si(3)–P(2) 113.66(2), Si(1)–O(1)–Si(2) 144.76(3), Si(1)–P(2)–Si(3) 96.89(2), Si(2)–P(1)–Si(3) 96.52(1). | ||
The distances from the phosphorus atoms to the silane entity Si(3) measure 224.6(1) and 224.5(1) pm, respectively. The bond lengths from the phosphorus atoms to the siloxane group are slightly shorter, with values between 220.8(1) and 220.2(1) pm. Similar bond lengths between phosphorus and silane or siloxane groups, respectively, can also be found in other complexes with silicon functionalized phosphorus atoms.6 The 31P{1H} NMR spectrum shows one signal at −266.5 ppm with one set of 29Si satellites with a coupling constant of 39.3 Hz. A second set of satellites cannot be observed. Compound 2 is not soluble in non- or weakly polar solvents. In THF, the coordinating DME ligands of the metal cation were substituted, so we used a mixture of DME and benzene-d6 for the NMR experiments, therefore, only 31P{1H} NMR spectra are available.
The reaction of barium hexamethyldisilazanide with 1 leads to a monomeric structure which is similar to the strontium derivative. [Ba{O(SiiPr2P)2SiiPr2}(dme)3] (3) crystallizes in the orthorhombic space group P212121 with one additional non-coordinated DME molecule per formula unit. In contrast to compound 2, the coordination sphere of the metal cation is completed by three DME molecules, giving a coordination number of eight (Fig. 3).8
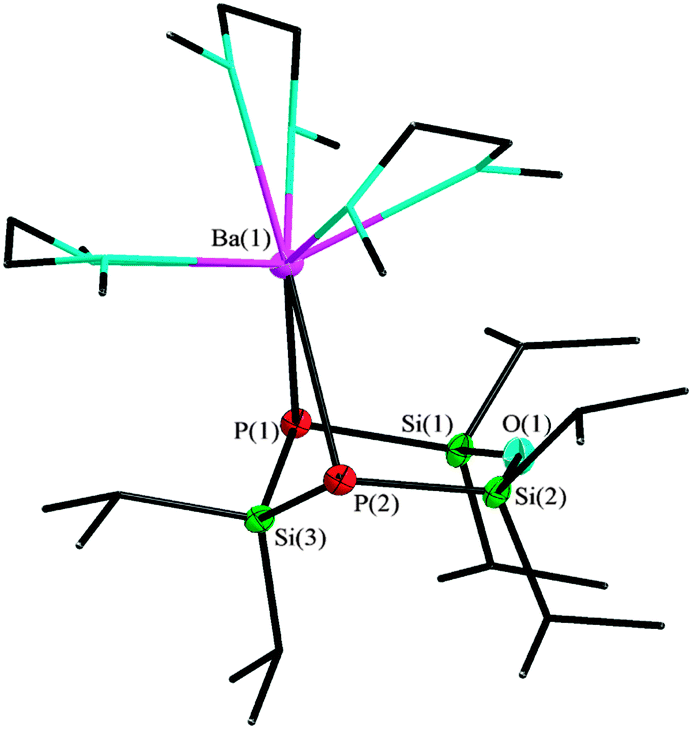 | ||
| Fig. 3 Molecular structure of 3, thermal ellipsoids represent a 50% probability level, hydrogen atoms of the organic groups are not shown. Selected atom distances [pm] and angles [°]: P(2)–Ba(1) 334.1(16), P(1)–Ba(1) 331.3(16), P(1)–Si(3) 223.2(20), P(2)–Si(1) 219.9(25), P(1)–Si(2) 219.9(27), Si(1)–O(1) 165.9(27); P(1)–Ba(1)–P(2) 68.38(4), P(1)–Si(3)–P(2) 113.39(8), Si(1)–P(2)–Si(3) 97.89(9), Si(1)–O(1)–Si(2) 139.41(33). | ||
Metalated cyclic silylphosphanides are suitable reactants for oxidative P–P bond formation by reaction with C2H4Br2.9,12 After a few minutes already, the reaction of 3 with 1,2-dibromoethane in benzene is completed and compound 4 can be detected by NMR analysis without occurrence of any phosphorus containing by-products (Scheme 3).10 Due to P–P bond formation, 4 is a bicyclic species, containing one three-membered P2Si and one five-membered OSi2P2 cyclic skeleton.
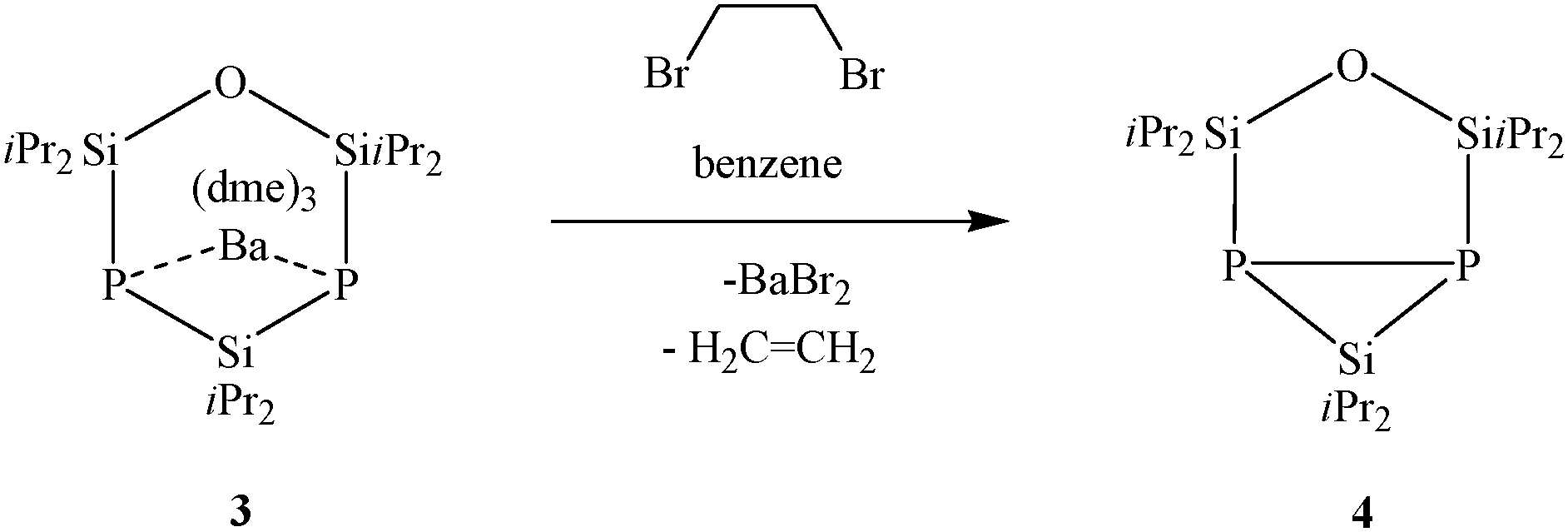 | ||
| Scheme 3 Oxidation of compound 3. | ||
In the 31P{1H} NMR spectrum, one observes one signal at −311.6 ppm, relating to the equivalent phosphorus atoms. The 29Si{1H} NMR spectrum shows one triplet at −20.6 ppm, belonging to the silane entity. This chemical shift is typical for three-membered P2Si heterocycles.5 Also the 29Si{1H} NMR signals of the siloxane entity confirm the monomeric structure of compound 4. This group appears as a multiplet at 37.1 ppm and represents the X part of an AA′X spin system (Fig. 4).11 An iterative analysis of the signal reveals a value of 1JSiP = 91 Hz and 2JSiP = −15 Hz for the silicon–phosphorus coupling and an approximate value of 50 Hz for the phosphorus–phosphorus coupling.
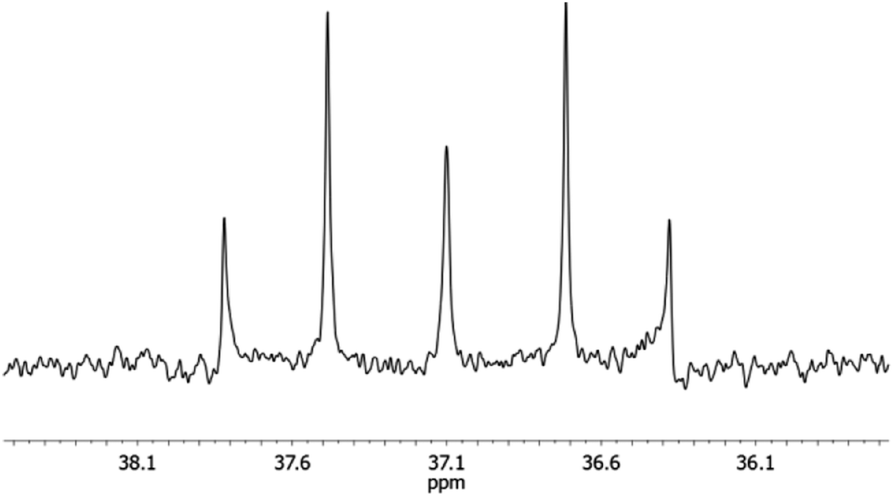 | ||
| Fig. 4 Expansion of the high resolution 29Si NMR spectrum of 4. | ||
While sterically unhindered three-membered P2Si heterocycles are usually unstable and easily react to give larger ring systems,5 compound 4 is highly stable towards temperature and UV light: neither exposition to 135 °C for a period of 18 h nor irradiation for 5 h (mercury vapour lamp) lead to any by-product. With the objective to confirm the molecular structure by X-ray analysis, we dissolved the bicyclic product 4 in toluene: after 7 days at −25 °C single crystals were obtained. Surprisingly, a different structure than the monomeric silylphosphine 4 was discovered.
The resulting compound 5 crystallizes in the monoclinic space group P21/n in form of colourless plates. The tricyclic product 5 has an asymmetric structure and can best be described as an isopropyl substituted P4Si2 cycle with two exocyclic siloxane entities, building one five-membered and one six-membered heterocycle each. Compound 5 is a dimer of 4, which can easily be identified in the siloxane bridged P(4)–Si(4)–P(3) fragment (Fig. 5). Therein, the previously present P–P bond in 4 is cleaved and now connects to a second entity of 4, precisely to the silane entity Si(3) and the phosphorus P(2).
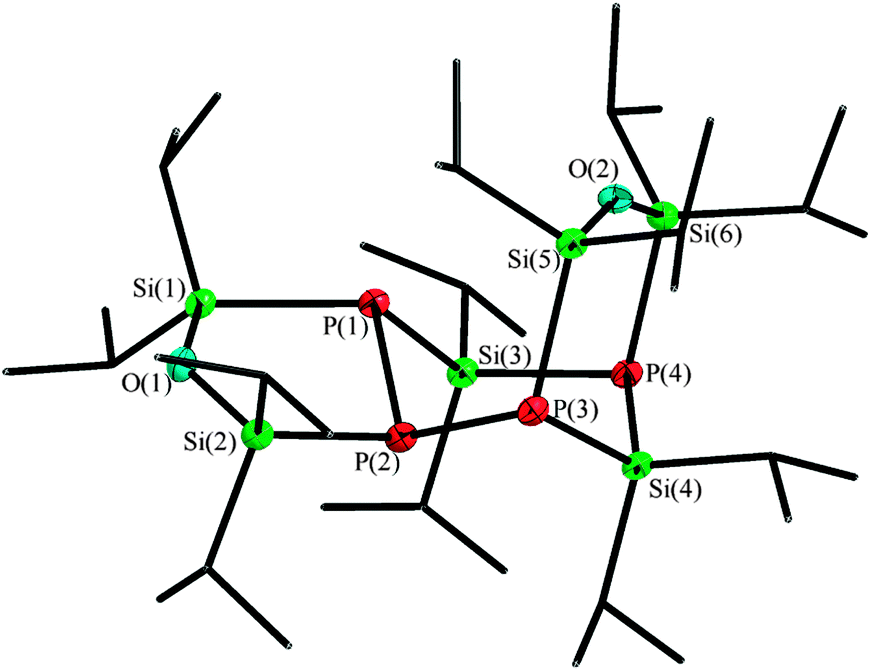 | ||
| Fig. 5 Molecular structure of 5, thermal ellipsoids represent a 50% probability level, hydrogen atoms of the organic groups are not shown. Selected atom distances [pm] and angles [°]: P(1)–P(2) 220.20(8), P(2)–P(3) 220.77(10), P(1)–Si(1) 230.63(10), P(4)–Si(4) 228.47(10), P(4)–Si(6) 228.75(9), Si(1)–O(1) 165.27(16); Si(1)–O(1)–Si(2) 131.90(11), Si(5)–O(2)–Si(6) 150.75(11), P(1)–Si(3)–P(4) 104.69(4), P(1)–P(2)–P(3) 110.16(4). | ||
The inner P4Si2 cycle shows a chair conformation, while the siloxane bridges are twisted against each other: their position is nearly orthogonal. With a value of 131.90(11)°, the Si(1)–O(1)–Si(2) angle is in the typical range of five-membered cyclic diphosphanyl-siloxanes.11 Compared to the metalated species of the six-membered cycle (compound 2 and 3), the Si(6)–O(2)–Si(5) angle of 150.75(11)° in compound 5 is more obtuse, which is due to the enlarged distance of the here non-coordinating phosphorus atoms P(3) and P(4). The silicon–phosphorus bond lengths, which vary between 228.47(10) pm for P(1)–Si(3) and 230.63(10) pm for Si(1)–P(1), accord to similar compounds.12 The 31P{1H} NMR spectrum shows three broad signals: P(1) and P(3) both appear at −214.3 ppm due to similar chemical environment, P(4) has a chemical shift of −265.5 ppm and P(2) can be identified at −161.0 ppm.
We furthermore observed that the dimerization depends on the nature of the solvent, since after extensive storage at −25 °C in DME decomposing reactions were observed instead. With the objective to comprehend the conditions of dimerization and decomposition better, we purified the monomeric product 4 by distillation in vacuo at oil bath temperatures of 200 °C (b.p. 96 °C, 1 × 10−3 mbar). After purification, the previous oily compound 4 is a white solid with a melting point of 83 °C. Interestingly, under consistent conditions as used before, neither dimerization nor decomposition of the pure diphosphasilirane 4 was observed. This led us to the conclusion that impurities like remaining metal bromides induce dimerization and decomposition, depending on the solvent used.
To sum up, cyclic silylphosphanes which contain two P–H units are valuable precursors for the synthesis of stable diphosphasiliranes. The bicyclic compound 4 presented here shows high stability towards temperature and UV light and can therefore be isolated via distillation. The subsequently observed oligomerization at low temperatures is promoted by impurities, since rearrangement is not observed for the purified compound 4.
The authors thank Dr E. Matern (Institut für Anorg. Chemie, KIT) for his valuable help with the NMR experiments and simulations. This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG).
Notes and references
- (a) W. W. Schoeller and T. Dabisch, J. Chem. Soc., Chem. Commun., 1985, 23, 1706 RSC; (b) W. W. Schoeller, V. Staemmler, P. Rademacher and E. Niecke, Inorg. Chem., 1986, 25, 4382 CrossRef CAS; (c) M. Weidenbruch, Chem. Rev., 1995, 95, 1479 CrossRef CAS; (d) M. Driess and H. Grützmacher, Angew. Chem., Int. Ed. Engl., 1996, 35, 828 CrossRef CAS.
- A. G. Baboul and H. B. Schlegel, J. Am. Chem. Soc., 1996, 118, 8444 CrossRef CAS.
- M. Baudler, J. Hahn, H. Dietsch and G. Fürstenberg, Z. Naturforsch., B, 1976, 31, 1305 Search PubMed.
- M. Driess, H. Pritzkow, S. Rell and R. Janoschek, Inorg. Chem., 1997, 36, 5212 CrossRef CAS.
- (a) M. Baudler and H. Jongebloed, Z. Anorg. Allg. Chem., 1979, 458, 9 CrossRef CAS; M. Baudler, Angew. Chem., 1982, 94, 520 Search PubMed; (b) M. Baudler, Angew. Chem., Int. Ed. Engl., 1982, 21, 492 CrossRef.
- (a) C. v. Hänisch and P. Kopecky, Z. Anorg. Allg. Chem., 2010, 636, 1522 CrossRef; (b) P. Kopecky, C. v. Hänisch, F. Weigend and A. Kracke, Eur. J. Inorg. Chem., 2010, 258 CrossRef CAS; (c) S. Traut, C. v. Hänisch and H.-J. Kathagen, Eur. J. Inorg. Chem., 2009, 777 CrossRef CAS; (d) C. von Hänisch and S. Stahl, Angew. Chem., 2006, 118, 2360 CrossRef.
- (a) G. Hoge, J. Am. Chem. Soc., 2004, 126, 9921 Search PubMed; (b) O. J. Scherer and R. Mergner, J. Organomet. Chem., 1972, 40, 64 CrossRef.
- Also the calcium congener [Ca{O(SiiPr2P)2SiiPr2}(dme)2] was obtained by analogous reaction of O(SiiPr2PH)2SiiPr2 (1) with calcium silazanide. Since the reaction showed only small yield, beside crystal structure determination no further analysis was possible.
- V. Cappello, J. Baumgartner, A. Dransfels and K. Hassler, Eur. J. Inorg. Chem., 2006, 4589 CrossRef CAS.
- With 1,2-dibromoethane the strontium derivative 2 reacts in an analogous manner as compound 3.
- R. Radeglia, J. Prakt. Chem., 1989, 331, 863 CrossRef.
- A. Kracke and C. v. Hänisch, Eur. J. Inorg. Chem., 2011, 3374 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and crystallographic data. CCDC 966215 (2), 966216 (3) and 966217 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02489c |
| This journal is © The Royal Society of Chemistry 2014 |