 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular N–H⋯Cl hydrogen bonds in the outer coordination sphere of a bipyridyl bisurea-based ligand stabilize a tetrahedral FeLCl2 complex†
Jesse V.
Gavette
a,
Christina M.
Klug
b,
Lev N.
Zakharov
c,
Matthew P.
Shores
*b,
Michael M.
Haley
*a and
Darren W.
Johnson
*a
aDepartment of Chemistry & Biochemistry and Materials Science Institute, University of Oregon, Eugene, OR 97403-1253, USA. E-mail: dwj@uoregon.edu; haley@uoregon.edu; Tel: +1-541-346-1253 Tel: +1-541-346-0456
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA. E-mail: matthew.shores@colostate.edu; Tel: +1-970-491-7235
cCAMCOR—Center for Advanced Materials Characterization in Oregon, University of Oregon, Eugene, OR 97403-1253, USA
First published on 15th May 2014
Abstract
A bipyridyl-based anion receptor is utilized as a ligand in a tetrahedral FeCl2 complex and demonstrates secondary coordination sphere influence through intramolecular hydrogen bonding to the chloride ligands as evidenced by X-ray crystallography.
Ligands capable of metal coordination while simultaneously hydrogen bonding to anionic guests have drawn considerable interest.1–4 Perhaps due to the ubiquity of iron and its relevance in biological systems, numerous coordination complexes designed as “hosts” for anions feature iron in their binding site. Some of these systems have utilized intramolecular hydrogen bonding to mimic the subtle secondary coordination sphere influences found in metalloproteins.5,6Intermolecular hydrogen bonding systems have also been used to allow secondary coordination of anions to modulate the spin states of metal complexes for potential applications in data storage and sensing.7–11 In light of these emerging applications and the inherent challenge in targeting coordination of a metal complex rather than just a single metal ion, the design of novel ligands that coordinate metal complexes through both direct chelation and hydrogen bonding, remains an active area of ion coordination chemistry.
Recently we reported anion binding studies of bipyridyl bisurea-based receptor 1 (Fig. 1).12 This receptor displayed a particular affinity toward the dihydrogen phosphate anion, H2PO4−, in 10% DMSO–chloroform-based solvent mixtures. The ditopic binding environment of this receptor, which provides two urea groups for convergent hydrogen bonding to stabilize negative charges and bipyridyl nitrogen lone-pairs for stabilizing positive charges, was the source of the preference toward this diprotic oxoanion. Based on this receptor design, it was recognized that the binding pocket should be suitable to a variety of guests possessing either (or both) positive and negative charges. Given the ubiquity of bipyridine as a ligand for transition metals, we were interested in investigating if the hydrogen bond donor groups on 1 would act in tandem with the bipyridyl group to coordinate and hydrogen bond to small metal complexes. Such molecular recognition of metal complexes has been termed “stereognostic” coordination chemistry.13
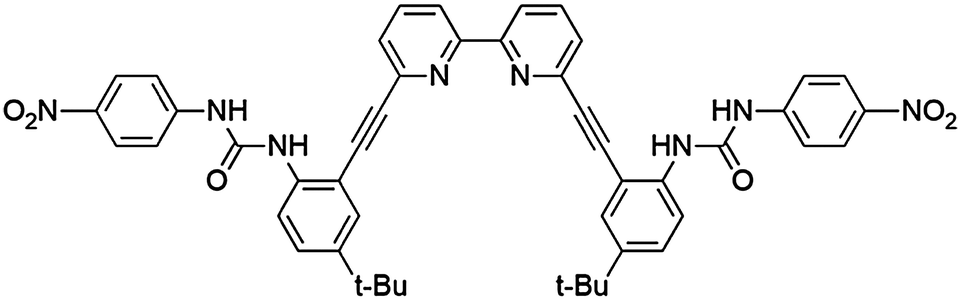 | ||
| Fig. 1 Structural representation of 1. | ||
Herein we report solid state X-ray analysis and SQUID studies of the coordination of 1 with FeCl2, indicating the formation of a tetrahedral 1·FeCl2 metal complex. Surprisingly, this species features four tight intramolecular hydrogen bonding interactions between the two urea groups and the coordinated chloride ligands remaining on the Fe2+ metal centre.
Slow evaporation of 1 with excess FeCl2·4H2O in acetonitrile resulted in yellow block-shaped crystals.14 The subsequent X-ray structure shows an Fe2+ ion coordinated in a tetrahedral geometry by two chlorides and the bipyridyl core; the binding is likely reinforced by the interaction between the two urea “arms” of the ligand and the metal-bound chloride ligands (Fig. 2a). The urea N–Cl bond distances and N–H⋯Cl bond angles indicate the formation of two moderate to weak hydrogen bonds15 to each of the coordinated chloride ligands by the respective urea functional groups (Table 1). Examples of this type of intramolecular hydrogen bonding to metal halides in the solid state are limited, and are typified by hydrogen bonding to a single halide ligand.16–19 The observation that this structure allows the formation of up to four intramolecular hydrogen bonds to the two coordinated halides on the metal highlights the novelty of the presented ligand design. Curiously, the average Fe–N and Fe–Cl distances of 1·FeCl2 (2.048(5) Å and 2.194(8) Å, respectively) are shorter on average than those of similarly structured biheteroaryl-coordinated tetrahedral FeCl2 complexes (with average distances ranging from 2.1029(65)–2.115(8) Å and 2.2209(6)–2.2303(7) Å, respectively).20–22 This may be a result of the ligand constricting the metal salt within the binding pocket. Additionally, the crystal structure reveals that the convergent coordination of the urea-based ligand appendages results in a helical twist in the binding conformation, and both enantiomers (P and M helices, Fig. 2b) are present in a racemic mixture in the solid state. Such guest-induced helical chirality has received much attention recently,23–25 and represents an interesting area of further research for this and related systems.
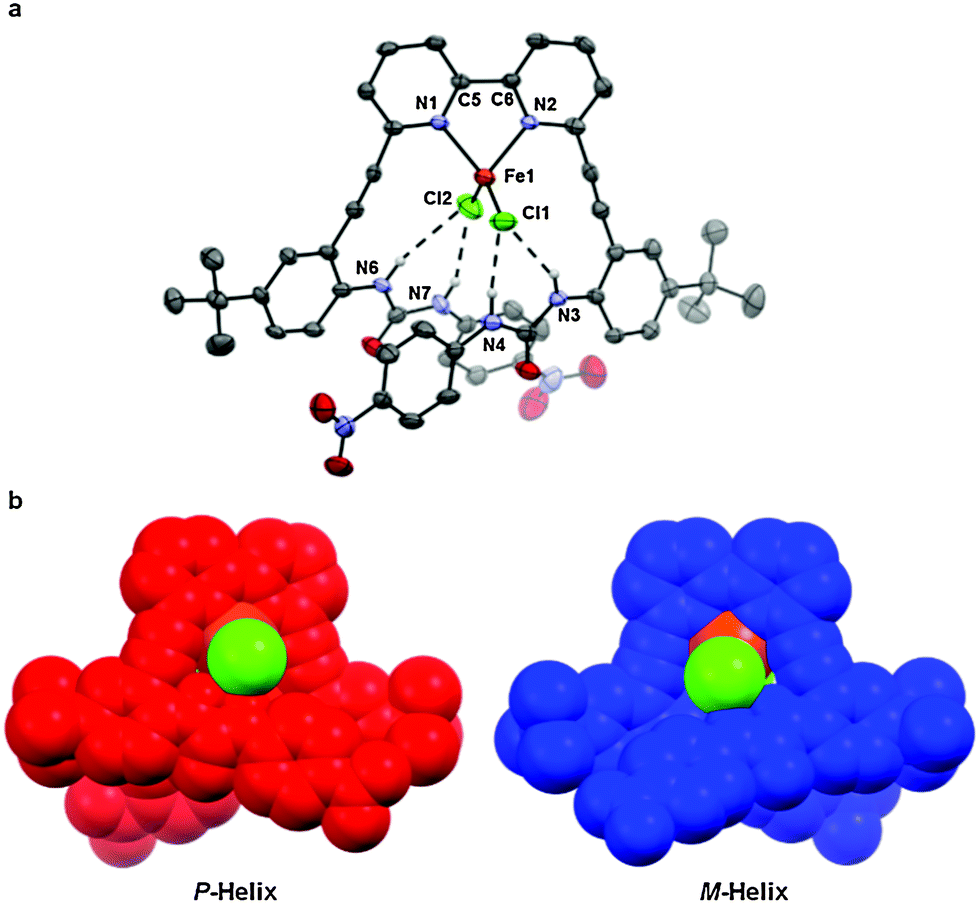 | ||
| Fig. 2 X-Ray data of 1 represented as (a) an ORTEP structure (ellipsoids at 50% probability and non-coordinating hydrogens removed for clarity) and (b) space filling structures of both P and M helices. | ||
| Bond | Distance | Angle |
|---|---|---|
| a Angle of N–H⋯Cl. | ||
| Fe1–N1 | 2.030(5) | — |
| Fe1–N2 | 2.066(5) | — |
| Fe1–Cl1 | 2.188(3) | — |
| Fe1–Cl2 | 2.201(3) | — |
| N3⋯Cl1 | 3.407(6) | 151a |
| N4⋯Cl1 | 3.096(7) | 156a |
| N6⋯Cl2 | 3.581(6) | 152a |
| N7⋯Cl2 | 3.190(6) | 171a |
| N1–Fe1–N2 | — | 79.6(2) |
| Cl1–Fe1–Cl2 | — | 122.06(10) |
| N1–Fe1–Cl1 | — | 112.23(17) |
| N2–Fe1–Cl1 | — | 110.64(18) |
| N1–Fe1–Cl2 | — | 108.94(17) |
| N2–Fe1–Cl2 | — | 115.45(16) |
In an effort to characterize the iron complex in solution, determination of the magnetic susceptibility (χM) by 1H NMR using Evans method26,27 was attempted. The low solubility of ligand 1 and of the resultant 1·FeCl2 complex in common organic solvents dictated the use of highly coordinating solvents (e.g. DMSO-d6) to achieve appreciable concentrations of the Fe2+ species in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 CDCl3:DMSO-d6 solution. The limited solubility and temperature range available in this solvent system lead to 1H NMR spectra of the metal complex that suffered from problems ranging from extreme peak broadening to spectra that were nearly identical to that of the free ligand. This prevented the collection of reliable magnetic susceptibility measurement values in solution. These observations are consistent with the dissociation of the metal salt and ligand in highly polar (competitive) solvents. The complex was also not stable under ESI-MS conditions, and dissociated iron species and free ligand represent the major ions observed, although small amounts of ligated Fe-complexes were observed (see Fig. S3 and S4, ESI†). The apparent dissociation in solution is likely perpetuated by a frustrated coordination environment indicated by the short Fe–N and Fe–Cl bond distances observed in the solid state.
:10 CDCl3:DMSO-d6 solution. The limited solubility and temperature range available in this solvent system lead to 1H NMR spectra of the metal complex that suffered from problems ranging from extreme peak broadening to spectra that were nearly identical to that of the free ligand. This prevented the collection of reliable magnetic susceptibility measurement values in solution. These observations are consistent with the dissociation of the metal salt and ligand in highly polar (competitive) solvents. The complex was also not stable under ESI-MS conditions, and dissociated iron species and free ligand represent the major ions observed, although small amounts of ligated Fe-complexes were observed (see Fig. S3 and S4, ESI†). The apparent dissociation in solution is likely perpetuated by a frustrated coordination environment indicated by the short Fe–N and Fe–Cl bond distances observed in the solid state.
Although virtually all tetrahedral Fe2+ complexes are high spin, the short Fe–N and Fe–Cl bond distances observed in 1·FeCl2 imply that the compound is close to the spin crossover regime. In the solid state, magnetic susceptibility measurements of 1·FeCl2 show a high spin Fe2+ (S = 2) species at 295 K with a χMT value of 3.78 emu K mol−1 (μeff = 5.5) (Fig. 3). This value is consistent with other reported high-spin tetrahedral Fe2+ complexes,20,22,28 supporting the tetrahedral structure observed by X-ray analysis. At lower temperatures, a slight decrease in susceptibility is observed, dropping to 3.15 emu K mol−1 (μeff = 5.0) at 15 K, with a more drastic decrease in the χMT value at 5 K. This downturn is consistent with zero-field splitting of the high-spin Fe2+ ion due to the low symmetry ligand field.
 | ||
| Fig. 3 Temperature dependence of χMT for 1·FeCl2, obtained under a 1000 Oe measuring field. Line added only as a guide for the eye. | ||
Coordination of bipyridyl-based ligands with steric bulk adjacent to the donor atom tends to allow spin crossover for octahedral Fe2+ complexes;29 otherwise, aromatic diimines generally lead to low-spin species. Meanwhile, chloride is a weak field ligand and often leads to high spin Fe2+ complexes. Since the Fe2+ ion in 1·FeCl2 is in a tetrahedral coordination environment, thermally accessible spin crossover would require that the ligand field imparted by 1 and two Cl− anions be significantly stronger than what the combination of bipyridyl and chloride typically generate in hexacoordinate complexes. This ligand field argument is consistent with the data shown in Fig. 3.
Another consideration is structural rigidity. The one tetrahedral Fe2+ species which undergoes spin crossover, [PhB(MesIm)3Fe-NdPPh3], has a more flexible ligand set imparted by a tris(carbene)borate and axial phosphoraniaminato ligands.30 These moieties allow for the proper ligand distortions necessary to observe spin state changes. For 1·FeCl2, we postulate these distortions are not favored due to the rigidity of the bipyridyl and the intramolecular hydrogen-bonding network. Thus, the complex remains trapped in the high-spin state even though the Fe–N bond lengths suggest that the low-spin state should be accessible.
In conclusion, we have demonstrated that a bipyridyl bisurea-based receptor designed to ditopically coordinate protic anions provides a suitable framework as a ligand toward metal halide salts. Solid state investigation of the Fe2+ complex reveals the presence of intramolecular hydrogen bonds between 1 and the metal-coordinated halide ligands. These findings demonstrate the potential of this and related systems31–35 to affect coordinated metal centres through non-covalent interactions. Additionally, the helical nature of the formed ligand complex presented offers a potential avenue for incorporating enantiospecific recognition into future generations of ligand design.
This work was supported by NIH grant R01-GM087398, which also funded early stage intellectual property that was licensed by SupraSensor Technologies, a company co-founded by D.W.J and M.M.H. M.P.S. and C.M.K. thank the NSF (CHE-1058889) and Colorado State University for support of this work.
Notes and references
- V. Amendola and L. Fabbrizzi, Chem. Commun., 2009, 513–531 RSC.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612–3620 RSC.
- J. Pérez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC.
- (a) A. S. Borovik, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed; (b) C. E. MacBeth, A. P. Golombek, V. G. Young, Jr., C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938–941 CrossRef CAS PubMed.
- (a) T. M. Dewey, J. Du Bois and K. N. Raymond, Inorg. Chem., 1993, 32, 1729–1738 CrossRef CAS; (b) A. S. Borovik, J. Du Bois and K. N. Raymond, Angew. Chem., Int. Ed. Engl., 1995, 34, 1359–1362 CrossRef CAS; (c) T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 CrossRef CAS.
- Z. Ni, A. M. McDaniel and M. P. Shores, Chem. Sci., 2010, 1, 615–621 RSC.
- C. M. Klug, A. M. McDaniel, S. R. Fiedler, K. A. Schulte, B. S. Newell and M. P. Shores, Dalton Trans., 2012, 41, 12577–12585 RSC.
- Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS PubMed.
- G. Lemercier, N. Bréfuel, S. Shova, J. A. Wolny, F. Dahan, M. Verelst, H. Paulsen, A. X. Trautwein and J.-P. Tuchagues, Chem. – Eur. J., 2006, 12, 7421–7432 CrossRef CAS PubMed.
- H. Z. Lazar, T. Forestier, S. A. Barrett, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Dalton Trans., 2007, 4276 RSC.
- J. V. Gavette, N. S. Mills, L. N. Zakharov, C. A. Johnson, D. W. Johnson and M. M. Haley, Angew. Chem., Int. Ed., 2013, 52, 10270–10274 CrossRef CAS PubMed.
- T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 CrossRef CAS.
- Crystallographic data for 1·FeCl2 (CCDC 978233): C48H42Cl2FeN8O6, M = 953.65, 0.12 × 0.07 × 0.05 mm, T = 173(2) K, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.666(9) Å, b = 11.764(9) Å, c = 18.282(14) Å, α = 78.585(16)°, β = 88.896(19)°, γ = 63.247(16)°, V = 2189(3) Å3, Z = 2, Dc = 1.447 Mg m−3, μ = 0.528 mm−1, F(000) = 988, 2θmax = 50.00°, 17504 reflections, 7664 independent reflections [Rint = 0.0971], R1 = 0.0838, wR2 = 0.1879 and GOF = 1.006 for 7664 reflections (586 parameters) with I > 2σ(I), R1 = 0.1693, wR2 = 0.2368 and GOF = 1.006 for all reflections, max/min residual electron density +1.126/−0.610 e Å3.
, a = 11.666(9) Å, b = 11.764(9) Å, c = 18.282(14) Å, α = 78.585(16)°, β = 88.896(19)°, γ = 63.247(16)°, V = 2189(3) Å3, Z = 2, Dc = 1.447 Mg m−3, μ = 0.528 mm−1, F(000) = 988, 2θmax = 50.00°, 17504 reflections, 7664 independent reflections [Rint = 0.0971], R1 = 0.0838, wR2 = 0.1879 and GOF = 1.006 for 7664 reflections (586 parameters) with I > 2σ(I), R1 = 0.1693, wR2 = 0.2368 and GOF = 1.006 for all reflections, max/min residual electron density +1.126/−0.610 e Å3. - J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley and Sons, 2nd edn, 2009 Search PubMed.
- T. E. Patten, C. Troeltzsch and M. M. Olmstead, Inorg. Chem., 2005, 44, 9197–9206 CrossRef CAS PubMed.
- D. Petrovic, M. Tamm and E. Herdtweck, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m217–m219 Search PubMed.
- C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400 RSC.
- C. M. Moore, D. A. Quist, J. W. Kampf and N. K. Szymczak, Inorg. Chem., 2014, 53, 3278–3280 CrossRef CAS PubMed.
- B. C. K. Chan and M. C. Baird, Inorg. Chim. Acta, 2004, 357, 2776–2782 CrossRef CAS.
- N. Rahimi, N. Safari, V. Amani and H. R. Khavasi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1370 CAS.
- O. Das, S. Chatterjee and T. K. Paine, JBIC, J. Biol. Inorg. Chem., 2013, 18, 401–410 CrossRef CAS PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC.
- H. Juwarker, J. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC.
- P. Prabhakaran, G. Priya and G. J. Sanjayan, Angew. Chem., Int. Ed., 2012, 51, 4006–4008 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- J. R. Hall, M. R. Litzow and R. A. Plowman, Aust. J. Chem., 1966, 19, 201–206 CrossRef CAS.
- Spin Crossover in Transition Metal Compounds I, ed. P. Gütlich and H. A. Goodwin, Springer, Berlin, Heidelberg, 2004, vol. 233 Search PubMed.
- J. J. Scepaniak, T. D. Harris, C. S. Vogel, J. Sutter, K. Meyer and J. M. Smith, J. Am. Chem. Soc., 2011, 133, 3824–3827 CrossRef CAS PubMed.
- C. A. Johnson, O. B. Berryman, A. C. Sather, L. N. Zakharov, M. M. Haley and D. W. Johnson, Cryst. Growth Des., 2009, 9, 4247–4249 CAS.
- C. N. Carroll, B. A. Coombs, S. P. McClintock, C. A. Johnson II, O. B. Berryman, D. W. Johnson and M. M. Haley, Chem. Commun., 2011, 47, 5539–5541 RSC.
- C. N. Carroll, O. B. Berryman, C. A. Johnson, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 2520–2522 RSC.
- M. M. Watt, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2013, 52, 10275–10280 CrossRef CAS PubMed.
- O. B. Berryman, C. A. Johnson, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2008, 47, 117–120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and magnetic measurements, X-ray analysis of receptor. CCDC 978233. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02297a |
| This journal is © The Royal Society of Chemistry 2014 |